Farmacocinética:
El levetiracetam es un compuesto altamente soluble y permeable. Su perfil farmacocinético es lineal e independiente del tiempo con baja variabilidad intra e intersujetos. No se presenta modificación de la depuración después de la administración de varias dosis. Tampoco hay evidencias relevantes de variabilidad relacionadas con el género, la raza o el ritmo circadiano. El perfil farmacocinético es comparable en voluntarios sanos y en pacientes con epilepsia.
Debido a su absorción completa y lineal, pueden predecirse los niveles plasmáticos en mg/kg de peso, a partir de una dosis de levetiracetam administrada por vía oral. Por lo que no es necesario monitorear los niveles plasmáticos de levetiracetam.
En niños y adultos se ha observado una correlación significativa entre las concentraciones plasmáticas y en saliva, de levetiracetam (la relación de concentración saliva/plasma se encuentra en el rango de 1 a 1.7 para las tabletas orales y 4 horas después de administrar la solución oral).
Adultos y Adolescentes
Absorción
El levetiracetam se absorbe rápidamente después de su administración oral. Su biodisponibilidad absoluta oral es cercana al 100%.
La concentración plasmática pico (Cmáx) se alcanza 1.3 h después de su administración. El estado estacionario se alcanza dos días después de iniciar la administración dos veces al día.
Las concentraciones pico (Cmáx) son típicamente 31 y 43 μg/ml, después de una administración de una dosis única de 1000 mg y la administración repetida de dosis de 1000 mg dos veces al día, respectivamente.
La tasa de absorción es independiente de la dosis y no se altera por la ingestión de alimentos.
Distribución
No se cuenta con datos disponibles de distribución en tejidos humanos.
Ni el levetiracetam ni su metabolito primario se unen significativamente a las proteínas plasmáticas (< 10%).
El volumen de distribución del levetiracetam es de aproximadamente 0.5 a 0.7 l/kg, un valor cercano al volumen corporal total de agua.
Biotransformación
El levetiracetam no es metabolizado extensamente en humanos. Su vía metabólica principal (24% de la dosis) es la hidrólisis enzimática del grupo acetamida. La producción del metabolito primario, ucb L057, no es mediada por las isoformas del citocromo P450 hepático. La hidrólisis del grupo acetamida fue cuantificable en gran número de tejidos, incluyendo a las células sanguíneas. El metabolito ucb L057 es farmacológicamente inactivo.
También se identificaron 2 metabolitos menores. Uno, obtenido por la hidroxilación del anillo pirrolidona (1.6% de la dosis) y el otro, por la apertura del anillo pirrolidona (0.9% de la dosis).
Otros compuestos no identificados participaron únicamente con el 0.6% de la dosis.
No se evidenció interconversión enantiomérica in vivo para el levetiracetam o para su metabolito principal.
En ensayos in vitro, el levetiracetam y su metabolito primario mostraron que no inhiben la actividad de las isoformas hepáticas principales del citocromo P450 (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 Y 1A2), de la glucuronil transferasa (UGT1A1y UGT1A6) y actividades de la epóxido hidroxilasa. Además, el levetiracetam no afecta la glucuronización in vitro del ácido valproico.
En cultivo de hepatocitos humanos, el levetiracetam tiene un efecto mínimo o nulo en la conjugación del etinilestradiol o de CYP1A1/2. Levetiracetam causó una inducción moderada de CYP2B6 y CYP3A4 a concentraciones elevadas (680 μg/mL); sin embargo, a concentraciones aproximadas a Cmáx, que se alcanza después de dosis repetidas de 1,500 mg/dos veces/día, los efectos no fueron considerados biológicamente relevantes. Debido a todo lo anterior, la interacción de levetiracetam con otras sustancias o viceversa es improbable.
Eliminación
La vida media plasmática en adultos es de 7 ± 1 horas y no varió con la dosis, vía de administración o administración repetida. El promedio de la depuración corporal total es de 0.96 mL/min/kg.
La principal vía de eliminación es la urinaria, con por lo menos el 95% de la dosis, (aproximadamente el 93% de la dosis se excretó en un periodo de 48 h). La excreción por vía fecal representa únicamente el 0.3% de la dosis.
La excreción urinaria acumulativa del levetiracetam y su metabolito primario durante las primeras 48 horas, representó el 66% y el 24% de la dosis, respectivamente.
La depuración renal de levetiracetam y de ucb L057 es de 0.6 y 4.2 mL/min/kg respectivamente, lo que indica que el levetiracetam es excretado por filtración glomerular con la subsecuente reabsorción tubular y que el metabolito primario también se excreta por secreción tubular activa además de la filtración glomerular. La eliminación de levetiracetam se correlaciona con la depuración de creatinina.
Grupos especiales de pacientes
Ancianos
En los ancianos, la vida media se incrementa en aproximadamente un 40% (10 a 11 h). Esto se relaciona con la disminución de la función renal en esta población.
Insuficiencia renal
La depuración corporal aparente de levetiracetam y su metabolito primario se correlaciona con la depuración de creatinina. Por lo que se recomienda ajustar la dosis diaria de mantenimiento de Keppra®, con base en la depuración de creatinina en pacientes con disfunción renal moderada y severa.
En pacientes anúricos con enfermedad terminal renal, la vida media fue de aproximadamente 25 y 3.1 h durante los periodos interdialítico e intradialítico, respectivamente.
La remoción fraccional de levetiracetam fue de 51% durante una sesión típica de diálisis de 4 horas.
Insuficiencia hepática
No hubo modificación relevante de la farmacocinética de levetiracetam en pacientes con insuficiencia hepática leve (Child-Pugh A) a moderada (Child-Pugh B). En sujetos con insuficiencia hepática severa (Child-Pugh C), la depuración total corporal fue del 50% que en sujetos normales, pero la depuración renal disminuida explica la mayor parte de la reducción.
No se necesita ajustar la dosis en pacientes con insuficiencia hepática leve a moderada. En pacientes con la disfunción hepática severa, la depuración de la creatinina puede subestimar la insuficiencia renal. Por lo tanto, se recomienda una reducción del 50% de la dosis diaria de mantenimiento cuando la depuración de la creatinina es < 60 mL/min/1.73m2.
Pacientes pediátricos
Lactantes y niños (de 1 mes a 4 años)
Tras la administración de dosis únicas (20 mg/kg) de solución oral 100 mg/mL a niños con epilepsia (de 1 mes a 4 años), levetiracetam fue rápidamente absorbido y los picos de concentraciones plasmáticas se observaron aproximadamente 1 hora tras la administración. Los resultados farmacocinéticos indicaron que la vida media era inferior (5.3 horas) que para adultos (7.2 horas) y el aclaramiento aparente era más rápido (1.5 mL/min/kg) que para adultos (0.96 mL/min/kg).
Niños (de 4 a 12 años)
Siguiendo un régimen de administración único (20 mg/ kg) en niños con diagnóstico de epilepsia, la vida media de levetiracetam es de 6.0 h. La depuración corporal aparente ajustado al peso fue de 1.43 ml/min/kg. Después de la administración repetida de dosis orales (20 a 60 mg/kg/día) a niños con epilepsia (de 4 a 12 años), el levetiracetam fue rápidamente absorbido. La concentración plasmática pico se observó de 0.5 a 1.0 hora después de su administración. Se observaron incrementos lineales y proporcionales a la dosis para la concentración plasmática pico y el área bajo la curva. La vida media de eliminación fue de aproximadamente 5 horas. La depuración corporal aparente fue de 1.1 mL/min/kg.
En el análisis farmacocinético de la población realizado en pacientes hasta 16 años de edad, el peso corporal se correlacionó significativamente con la depuración aparente (la depuración aumentó con un incremento en el peso corporal) y con el volumen aparente de distribución. La edad también tuvo una influencia en ambos parámetros. Este efecto fue pronunciado para los infantes más pequeños, y bajó al ir aumentado la edad, para volverse imperceptible alrededor de los 4 años de edad.
En ambos análisis farmacocinéticos de la población, hubo aproximadamente un aumento del 20% de la depuración aparente del levetiracetam cuando fue coadministrado con un fármaco antiepiléptico inductor de enzima.
Farmacodinamia:
Grupo Farmacoterapéutico
Antiepiléptico, código ATC: N03AX14.
La sustancia activa, levetiracetam, es un derivado de la pirrolidona (S-enantiómero de α-etil-2-oxo-1-pirrolidin acetamida), no relacionado químicamente con las sustancias activas antiepilépticas existentes.
Mecanismo de acción
El mecanismo de acción del levetiracetam no ha sido totalmente dilucidado, pero parece ser diferente a los mecanismos de los fármacos antiepilépticos actuales. Los experimentos in vivo e in vitro sugieren que el levetiracetam no altera las características celulares básicas y la neurotransmisión normal.
Los estudios in vitro muestran que el levetiracetam afecta los niveles de Ca2+ intraneuronal por inhibición parcial de las corrientes de Ca2+ de tipo N y por reducción de la liberación de Ca2+ de las reservas intraneuronales. Adicionalmente, revierte parcialmente las reducciones en las corrientes dependientes de GABA y glicina inducidas por zinc y β-carbolinas. Más aún, el levetiracetam ha mostrado en estudios in vitro que se une a sitios específicos del tejido cerebral de los roedores. Este sitio de unión es la proteína de la vesícula sináptica 2A, que se cree está involucrada en la fusión de las vesículas y liberación de neurotransmisores. El levetiracetam y los análogos relacionados muestran un rango de afinidad para la unión de la proteína de la vesícula sináptica 2A que se correlaciona con la potencia de su protección anticonvulsiva en el modelo audiogénico de epilepsia del ratón. Este hallazgo sugiere que la interacción entre levetiracetam y la proteína de la vesícula sináptica 2A parece contribuir al mecanismo de acción antiepiléptico del fármaco.
Efectos farmacodinámicos
El levetiracetam induce protección contra las crisis en un amplio intervalo de modelos animales de crisis generalizadas primarias y parciales (focales) sin tener efecto pro-convulsivo. Su metabolito principal es inactivo.
En el hombre, la actividad en las condiciones de epilepsias parciales (focales) y generalizadas (descargas epileptiformes/ respuestas fotoparoxísticas) han confirmado el amplio espectro de su perfil farmacológico preclínico.
ESTUDIOS CLÍNICOS
Crisis parciales (focales) en adultos y adolescentes de 16 años de edad con epilepsia.
Monoterapia
El estudio de monoterapia fue diseñado como una comparación de no-inferioridad, doble ciego y de grupos paralelos de levetiracetam y carbamazepina de liberación controlada en pacientes de 16 años de edad y mayores con epilepsia de diagnóstico nuevo o reciente. Las crisis convulsivas eran crisis parciales (focales) no provocadas (tipo IA, IB o IC de origen focal claro) o crisis convulsivas tónico-clónicas generalizadas (sin origen focal claro), clasificables de acuerdo con la Clasificación Internacional de Crisis Epilépticas. El estudio fue realizado en 85 centros de 13 países (Europa y Sudáfrica).
Al final del periodo de exploración de una semana, los pacientes elegibles fueron estratificados por tipo de crisis convulsiva (IA, IB/IC o IC/IIE sin origen focal claro) y fueron asignados aleatoriamente para recibir CBZ CR (N = 291) o LEV (N = 285) hasta por 121 semanas dependiendo de la respuesta. Desde un punto de vista conservador, se utilizó una formulación de carbamazepina de liberación controlada (CR) para minimizar las reacciones adversas.
El tratamiento fue iniciado con una titulación de hasta 2 semanas con carbamazepina CR, 200 mg/ día o levetiracetam, 500 mg/día, después de una semana de estabilización con el nivel de dosis blanco 1 (carbamazepina CR, 400 mg/día o levetiracetam, 1000 mg/día).
Los pacientes debían permanecer con este nivel de dosis durante 26 semanas (periodo de evaluación), siempre y cuando no presentaran ataques, seguido por un periodo de 26 semanas adicionales de terapia de mantenimiento. Si un paciente tenía una crisis convulsiva durante el periodo de evaluación, se hacía un escalonamiento (durante dos semanas con una semana de estabilización) de la dosis blanco al nivel 2 (carbamazepina CR, 800 mg/día o levetiracetam, 2000 mg/día). De forma similar, los pacientes que presentaron un ataque durante el periodo de evaluación del nivel de dosis 2 podían ser sometidos a un posterior ajuste de dosis de carbamazepina, 1200 mg/día o levetiracetam, 3000 mg/día. En los niveles de dosis 2 y 3, el periodo de evaluación continuó durante 26 semanas seguido por un periodo de mantenimiento de 26 semanas.
579 pacientes fueron asignados aleatoriamente. Aproximadamente la mitad de los pacientes de cada grupo de tratamiento completaron el estudio (53.6% de los pacientes asignados aleatoriamente al grupo CBZ y 54.0% de los pacientes asignados aleatoriamente el grupo LEV), porcentaje similar entre los dos grupos de tratamiento. La distribución por categoría de tipo de ataque fue similar en ambos grupos de tratamiento, y aproximadamente el 86.7% de los pacientes fue clasificado como paciente con ataque parcial de origen focal claro. La mayoría de los pacientes se mantuvo en un nivel de dosis 1 (81.7% de los pacientes asignados aleatoriamente a CBZ y 73.4% de los pacientes asignados aleatoriamente a LEV en la población PP).
El criterio de evaluación primario definido prospectivamente fue la proporción de pacientes de la población PP con un periodo libre de crisis convulsivas de 6 meses con la última dosis evaluada.
173 (73.0%) pacientes de la PP del grupo LEV estuvieron libres de ataques durante al menos 6 meses con la última dosis evaluada, en comparación con 171 pacientes (72.8%) del grupo CBZ. La diferencia absoluta ajustada entre los grupos LEV y CBZ (IC del 95% de dos colas) obtenida a partir de un modelo de regresión logística, incluyendo un factor para la categoría del tipo de crisis convulsiva evaluada al final (IA/IB/IC versus IC/IIE) igual a 0.2% (-7.8%; 8.2%). El límite inferior del intervalo de confianza (-7.8%) estuvo por encima del límite de no-inferioridad establecido en el protocolo (-15%) para este análisis de eficacia primario, por lo que LEV puede ser considerado como no inferior para CBZ en la proporción de pacientes sin crisis convulsivas durante al menos 6 meses con la primera dosis evaluada en la población PP. Al considerar los otros criterios de evaluación clínicamente relevantes, el 56.6% y el 58.5% de los pacientes de LEV y CBZ, respectivamente, no presentaron crisis convulsivas durante 1 año.
Terapia adyuvante
La eficacia de levetiracetam como terapia adyuvante (adicional a otros medicamentos antiepilépticos) en adultos fue establecida en tres estudios clínicos multicéntricos, aleatorizados, doble ciego y controlados con placebo en pacientes con crisis refractarias de aparición parcial (focal) con o sin generalización secundaria. En todos estos estudios se utilizó la formulación del comprimido. En estos estudios, 904 pacientes fueron asignados aleatoriamente al grupo placebo, 1000 mg, 2000 mg o 3000 mg/día. Los pacientes reclutados en el Estudio 1 o Estudio 2 habían tenido crisis refractarias de aparición parcial (focal) durante al menos dos años y habían tomado dos o más AEDs clásicos. Los pacientes reclutados en el Estudio 3 habían tenido crisis refractarias de aparición parcial (focal) durante al menos un año y habían tomado un AED conocido. Al momento del estudio, los pacientes estaban tomando un régimen de dosis estable al menos de un AED (y máximo dos). Durante el periodo basal, los pacientes tenían que haber presentado al menos dos crisis de aparición parcial (focal) durante cada periodo de 4 semanas.
Estudio 1
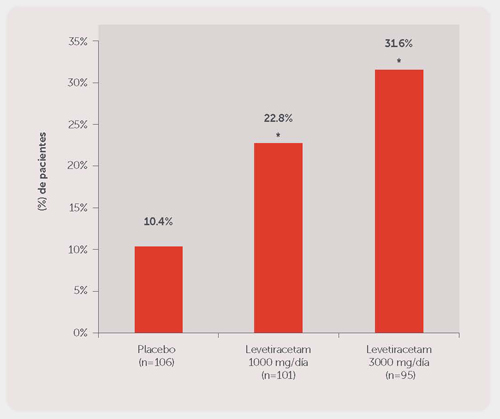
El Estudio 1 fue un estudio doble ciego, controlado con placebo y de grupos paralelos realizado en 41 centros de Estados Unidos, en comparación con levetiracetam, 1000 mg/día (N = 98), levetiracetam, 3000 mg/día (N = 101) y placebo (N = 95) administrado en dosis divididas en partes iguales dos veces al día. Después del periodo basal prospectivo de 12 semanas, los pacientes fueron asignados aleatoriamente a uno de los tres grupos de tratamiento descritos anteriormente. El periodo de tratamiento de 18 semanas estuvo compuesto de un periodo de ajuste de 6 semanas, seguido por un periodo de evaluación de dosis fija de 12 semanas, durante el cual los regímenes de AED concomitantes se mantuvieron constantes. La medida de eficacia primaria fue una comparación entre grupos de la reducción porcentual en la frecuencia de crisis parciales (focales) con relación al placebo durante todo el periodo de tratamiento (tomando en cuenta las últimas 2 semanas de ajuste + 12 semanas del periodo de evaluación). Las variables de los resultados secundarios incluyeron la tasa del respondedor (incidencia de pacientes con ≥ 50% de reducción desde el estado basal en la frecuencia de crisis de aparición parcial (focal)). Los resultados del análisis del Estudio 1 se presentan en la Tabla 1.
Tabla 1: Reducción en la media del placebo en la frecuencia semanal de crisis de aparición parcial (focal) en el Estudio 1
| Placebo
(N = 95) | Levetiracetam 1000 mg/día
(N = 98) | Levetiracetam 3000 mg/día
(N = 101) |
| n | 93 | 94 | 98 |
| Porcentaje de la reducción en la frecuencia de crisis parciales (focales) sobre el placebo | --- | 20.9%* | 27.7%* |
*P < 0.001
En la Figura 1 se presenta el porcentaje de pacientes (eje y) que alcanzó una reducción ≥50% en las tasas de crisis semanales desde el estado basal en la frecuencia de crisis de aparición parcial (focal) durante el periodo de tratamiento (tomando en cuenta las últimas 2 semanas de ajuste + 12 semanas del periodo de evaluación) dentro de los tres grupos de tratamiento (eje x).
Figura 1. Tasa de Respondedor (≥50% de reducción desde el estado basal) en el Estudio 1
*P < 0.001 versus placebo (regresión logística).
Estudio 2
El Estudio 2 fue un estudio doble ciego, controlado con placebo y transversal realizado en 62 centros de Europa en el que se comparó levetiracetam, 1000 mg/día (N = 106), levetiracetam, 2000 mg/día (N = 106) y placebo (N = 112) administrado en dosis iguales dos veces al día.
El primer periodo del estudio (Periodo A) fue diseñado para ser analizado como un estudio de grupos paralelos. Después de un periodo basal prospectivo de hasta 12 semanas, los pacientes fueron asignados aleatoriamente a uno de los tres grupos de tratamiento descritos anteriormente. El periodo de tratamiento de 16 semanas estuvo compuesto de un periodo de ajuste de 4 semanas seguido por un periodo de evaluación de dosis fija de 16 semanas durante el cual los regímenes de AEDs concomitantes se mantuvieron constantes. La medida de eficacia primaria fue una comparación entre grupos de la reducción porcentual en la frecuencia semanal de crisis parciales (focales) con relación al placebo durante todo el periodo de tratamiento. Las variables de los resultados secundarios incluyeron la tasa del respondedor (incidencia de pacientes con ≥ 50% de reducción desde el estado basal en la frecuencia de crisis de aparición parcial (focal)). Los resultados del análisis del Periodo A se presentan en la Tabla 2.
Tabla 2: Reducción en la media del placebo en la frecuencia semanal de crisis de aparición parcial (focal) en el Estudio 2: Periodo A
| Placebo
(N = 112) | Levetiracetam 1000 mg/día
(N = 106) | Levetiracetam 2000 mg/día
(N = 106) |
| n | 106 | 101 | 95 |
| Porcentaje de la reducción en la frecuencia de crisis parciales (focales) sobre el placebo | --- | 16.4%* | 17.7%** |
*P = 0.006, **P=0.003
En la Figura 2 se presenta el porcentaje de pacientes (eje y) que alcanzó una reducción ≥50% en las tasas de crisis semanales desde el estado basal en la frecuencia de crisis de inicio parcial (focal) durante el periodo de tratamiento dentro de los tres grupos de tratamiento (eje x).
Figura 2. Tasa de Respondedor (≥50% de reducción desde el estado basal) en el Estudio 2: Periodo A
*P = 0.019, **P<0.001 versus placebo (regresión logística).
Estudio 3
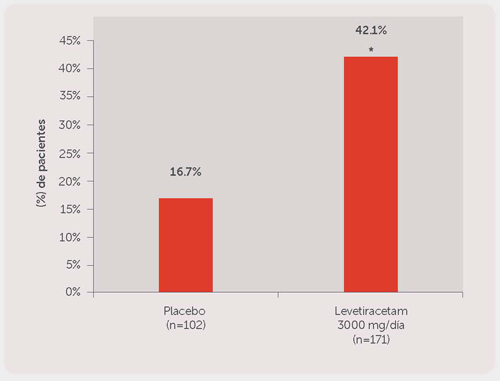
El Estudio 3 fue un estudio doble ciego, controlado con placebo y de grupos paralelos, realizado en 47 centros de Europa en el que se comparó levetiracetam, 3000 mg/día (N = 181) y placebo (N = 105) administrado a pacientes con crisis refractarias de aparición parcial (focal), con o sin generalización secundaria, que únicamente recibieron un AED concomitante. El medicamento de estudio fue administrado en dos dosis divididas. Después del periodo basal prospectivo de 12 semanas, los pacientes fueron asignados aleatoriamente a uno de los dos grupos de tratamiento descritos anteriormente. El periodo de tratamiento de 16 semanas consistió en un periodo de ajuste de 4 semanas, seguido por un periodo de evaluación adicional de dosis fija de 12 semanas y un período de 2 semanas del período de evaluación de los pacientes con respuesta durante el cual los regímenes de AEDs concomitantes se mantuvieron constantes. La medida de eficacia primaria fue una comparación entre grupos de la reducción porcentual en la frecuencia semanal de crisis parciales (focal) con relación al placebo durante todo el periodo de tratamiento aleatorizado adicional (teniendo en cuenta 12 semanas de ajuste adicional + 2 semanas del periodo de evaluación de los pacientes con respuesta). Las variables de los resultados secundarios incluyeron la tasa del respondedor (incidencia de pacientes con ≥50% de reducción desde el estado basal en la frecuencia de crisis de aparición parcial (focal)). Los resultados del análisis del Estudio 3 se presentan en la Tabla 3.
Tabla 3: Reducción en la media del placebo en la frecuencia semanal de crisis de aparición parcial (focal) en el Estudio 3.
| Placebo
(N = 105) | Levetiracetam 3000 mg/día
(N = 181) |
| n | 102 | 171 |
| Porcentaje de la reducción en la frecuencia de crisis parciales (focales) sobre el placebo | --- | 22.2% |
*P < 0.001
En la Figura 3 se presenta el porcentaje de pacientes (eje y) que alcanzó una reducción ≥50% en las tasas de crisis semanales desde el estado basal en la frecuencia de crisis de inicio parcial (focal) durante el periodo de tratamiento adicional (tomando en cuenta las 12 semanas adicionales de evaluación + 2 semanas periodo de evaluación de pacientes con respuesta) dentro de los dos grupos de tratamiento (eje x).
Figura 3. Tasa de Respondedor (≥50% de reducción desde el estado basal) en el Estudio 3.
*P<0.001 versus placebo (regresión logística).
En un análisis combinado, el porcentaje de pacientes que alcanzaron una reducción de 50% o mayor desde el estado basal en la frecuencia de crisis de aparición parcial (focal) por semana con una dosis estable (12/14 semanas) fue de 27.7%, 31.6% y 41.3% de pacientes con 1000, 2000 o 3000 mg de levetiracetam, respectivamente y de 12.6% en pacientes con placebo.
Crisis parciales (focales) en pacientes pediátricos con epilepsia
Pacientes pediátricos (4 a 16 años de edad)
La eficacia de levetiracetam como terapia adyuvante (adicional a otros medicamentos antiepilépticos) en pacientes pediátricos fue establecida en un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo realizado en 60 centros de Norteamérica, en niños de 4 a 16 años de edad con crisis parciales (focales) no controladas con medicamentos antiepilépticos estándar (AEDs). Los pacientes elegibles con una dosis estable de 1-2 AEDs (fármacos antiepilépticos), que seguían presentando al menos cuatro crisis de aparición parcial (focal) durante las 4 semanas anteriores a la exploración, así como al menos 4 crisis de aparición parcial (focal) en cada uno de los periodos basales de 4 semanas, fueron asignados aleatoriamente para recibir levetiracetam o placebo. El estudio incluyó a 198 pacientes (levetiracetam N = 101, placebo N = 97) con crisis refractarias de aparición parcial (focal), con o sin generalización secundaria. El estudio consistió en un periodo basal de 8 semanas y un periodo de ajuste de 4 semanas seguido por un periodo de evaluación de 10 semanas. La administración de levetiracetam se inició con una dosis de 20 mg/kg/día en dos dosis divididas. Durante el periodo de tratamiento, las dosis de levetiracetam fueron ajustadas en incrementos de 20 mg/kg/día, en intervalos de 2 semanas hasta la dosis blanco de 60 mg/kg/día. La medida de eficacia primaria fue una comparación entre grupos de la reducción porcentual en la frecuencia semanal de crisis parciales (focales) con relación al placebo durante todo el periodo de tratamiento de 14 semanas (ajuste + periodo de evaluación). Las variables de los resultados secundarios incluyeron la tasa del respondedor (incidencia de pacientes con ≥ 50% de reducción desde el estado basal en la frecuencia semanal de crisis de aparición parcial (focal)). La Tabla 4 presenta los resultados de este estudio.
Tabla 4: Reducción en la media del placebo en la frecuencia semanal de crisis de aparición parcial (focal)
| Placebo
(N = 97) | Levetiracetam
(N = 101) |
| Porcentaje de la reducción en la frecuencia | --- | 26.8%* |
| de crisis parciales (focales) sobre el placebo | | |
*P = 0.0002
En la Figura 4 se presenta el porcentaje de pacientes (eje y) que alcanzó una reducción ≥50% en las tasas de crisis semanales desde el estado basal en la frecuencia de crisis de aparición parcial (focal) durante el periodo de tratamiento aleatorizado total (ajuste + periodo de evaluación) dentro de los dos grupos de tratamiento (eje x).
Figura 4. Tasa de Respondedor (≥50% de reducción desde el estado basal)
*P = 0.0002 versus placebo (regresión logística).
Con un tratamiento a largo plazo continuo, el 11.4% de los pacientes estuvieron libres de crisis al menos durante 6 meses y 7.2% estuvieron libres de crisis al menos durante un año.
Pacientes pediátricos (1 mes a 4 años de edad)
La eficacia de levetiracetam como terapia de adición en pacientes pediátricos fue establecida en un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo realizado en niños de 1 mes a menos de 4 años de edad con crisis de inicio parcial (focal) no controladas con medicamentos antiepilépticos estándar (AEDs). Los pacientes elegibles con una dosis estable de 1-2 AEDs, que presentaron al menos dos crisis de inicio parcial (focal) durante un video - electroencefalograma (EEG) basal a 48 horas fueron asignados aleatoriamente para recibir levetiracetam o placebo. El estudio incluyó a 116 pacientes (levetiracetam N = 60, placebo N = 56) con crisis refractarias de inicio parcial (focal), con o sin generalización secundaria. La asignación aleatoria fue estratificada por rango de edad de la siguiente forma: 1 mes a menos de 6 meses de edad (levetiracetam N = 4, placebo = 4), 6 meses a menos de 1 año de edad (levetiracetam N = 8, placebo = 7), 1 año a menos de 2 años de edad (levetiracetam N = 20, placebo = 18) y 2 años a menos de 4 años de edad (levetiracetam N = 28, placebo = 27). El estudio consistió en un periodo de tratamiento de 5 días que incluyó un periodo de ajuste de un día seguido por un periodo de mantenimiento de 4 días. La dosis fue determinada por edad y peso de la siguiente forma: los niños de 1 mes a menos de 6 meses de edad fueron asignados a una dosis objetivo de 40 mg/kg/día y los niños de 6 meses a menos de 4 años de edad fueron asignados aleatoriamente a una dosis objetivo de 50 mg/kg/día. La medida principal de la efectividad fue la tasa de respondedores (porcentaje de pacientes con una reducción de ≥ 50% en promedio de la frecuencia de las crisis de inicio parcial (focal) diarias desde el inicio) evaluada por medio de un lector central ciego utilizando un lector central cegado usando EEG video por 48 horas. Un total de 109 pacientes fueron incluidos en el análisis de eficacia. Se observó una diferencia estadísticamente significativa entre el medicamento y el placebo (Figura 5). El efecto del tratamiento asociado con levetiracetam fue consistente entre los grupos de edad.
Figura 5. Tasa de Respondedores para todos los pacientes de 1 mes a < 4 años de edad (≥ 50% de reducción desde el estado basal)
*Estadísticamente significativo versus placebo.
Crisis mioclónicas en pacientes ≥ 12 años de edad
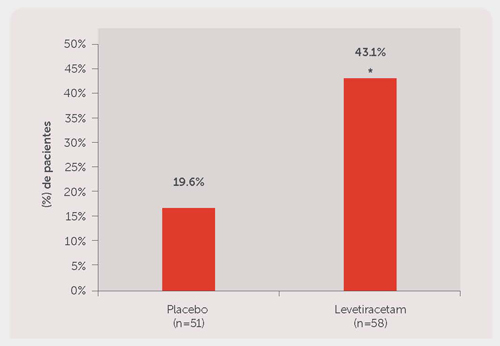
La eficacia de levetiracetam como terapia adyuvante (adicional a otros medicamentos antiepilépticos) en pacientes de 12 años de edad y mayores con epilepsia mioclónica juvenil con crisis mioclónicas fue establecida en un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo realizado en 37 centros de 14 países. Los pacientes elegibles con una dosis estable de 1 AED, que presentaban uno o dos crisis mioclónicas por día al menos durante 8 días durante el periodo basal prospectivo de 8 semanas fueron asignados aleatoriamente para recibir levetiracetam o placebo. El estudio incluyó a 121 pacientes (levetiracetam N = 61, placebo N = 60) con epilepsia idiopática generalizada, incluyendo epilepsia mioclónica juvenil, epilepsia juvenil con ausencias o epilepsia con crisis tónico-clónicas generalizadas al despertar. La mayoría de los pacientes tenían epilepsia mioclónica juvenil. Los pacientes fueron ajustados durante 4 semanas hasta una dosis blanco de 3000 mg/día y fueron tratados con una dosis estable de 3000 mg/día durante 12 semanas (periodo de evaluación). El medicamento de estudio fue administrado en dos dosis divididas.
La medida de eficacia primaria fue la proporción de pacientes con al menos un 50% de reducción en el número de días por semana con una o más crisis mioclónica durante el periodo de tratamiento (ajuste + periodo de evaluación), en comparación con el estado basal. Las variables secundarias incluyeron ausencia de crisis (mioclónicas) y la tasa de respondedores en la frecuencia de crisis mioclónicas por semana durante el periodo de tratamiento. La Tabla 5 presenta los resultados de este estudio.
Tabla 5: Tasa de respondedores (≥50% de reducción desde el estado basal) en días de crisis mioclónicas por semana
| Placebo
(N = 60) | Levetiracetam
(N = 61) |
| n | 60 | 60 |
| Porcentaje de respondedores | 23.3% | 58.3%* |
| Odds-Ratio (índice de probabilidad) (Lev vs. PBO) | | 4.77* |
*P = 0.0002, basada en el modelo logístico de regresión.
Con el tratamiento continuo a largo plazo, el 28.6% de los pacientes estuvieron libres de crisis mioclónicas durante al menos 6 meses y 21.0% estuvieron libres de crisis mioclónicas al menos durante un año.
Crisis tónico-clónicas generalizadas primarias en pacientes ≥6 años de edad
La eficacia de levetiracetam como terapia adyuvante (adicional a otros medicamentos antiepilépticos) en pacientes de 6 años de edad y mayores con epilepsia generalizada idiopática con crisis tónico-clónicas generalizadas primaras (PGTC por sus siglas en inglés) fue establecida en un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo realizado en 50 centros de 8 países. Los pacientes elegibles con una dosis estable de 1-2 AEDs con al menos tres crisis PGTC durante el periodo basal combinado de 8 semanas (al menos una crisis LGTC durante las 4 semanas previas al periodo basal prospectivo y al menos una crisis PGTC durante el periodo basal prospectivo de 4 semanas) fueron asignados aleatoriamente a levetiracetam o placebo. El periodo basal combinado de 8 semanas se refiere a “basal” en el resto de esta sección. El estudio incluyó a 164 pacientes (levetiracetam N = 80, placebo N = 84) con epilepsia idiopática generalizada (predominantemente epilepsia mioclónica juvenil, epilepsia juvenil con ausencias, epilepsia infantil con ausencias o epilepsia con Gran Mal al despertar) presentó crisis tónico-clónicas generalizadas primarias. Cada uno de estos síntomas de epilepsia idiopática generalizada estaba representado en esta población de pacientes. La dosis de los pacientes fue ajustada durante 4 semanas a una dosis blanco de 3000 mg/ día en adultos o una dosis blanco pediátrica de 60 mg/ kg/día y los pacientes fueron tratados con una dosis estable de 3000 mg/día (o 60 mg/kg en niños) durante 20 semanas (periodo de evaluación). El medicamento de estudio fue administrado en dos dosis iguales al día.
La medida de eficacia primaria fue el porcentaje de reducción desde el estado basal en la frecuencia semanal de PGTC en los grupos de levetiracetam y placebo durante el periodo de tratamiento (ajuste + periodo de evaluación). Hubo una disminución estadísticamente significativa desde el estado basal en la frecuencia PGTC en los pacientes tratados con levetiracetam en comparación con los pacientes tratados con placebo. La significancia estadística versus placebo indica un valor p de <0.05.
Tabla 6: Mediana del porcentaje de reducción desde el estado basal en la frecuencia de crisis PGTC por semana
| Placebo
(N = 84) | Levetiracetam
(N = 80) |
| n | 84 | 78 |
| Porcentaje de la reducción en la frecuencia de crisis PGTC | --- | 28.3%* |
*p=0.004
En la Figura 6 se presenta el porcentaje de pacientes (eje y) que alcanzó una reducción ≥50% en las tasas de crisis semanales desde el estado basal en la frecuencia de crisis PGTC durante el periodo de tratamiento aleatorizado total (ajuste + periodo de evaluación) dentro de los dos grupos de tratamiento (eje x).
Figura 6. Tasa de Respondedor (≥50% de reducción desde el estado basal) en la frecuencia de crisis PGTC por semana
*P < 0.001 versus placebo (regresión logística).
Con un tratamiento a largo plazo continuo, el 47.4% de los pacientes estuvieron libres de crisis tónico-clónicas al menos durante 6 meses y 31.5% estuvieron libres de crisis tónico-clónicas al menos durante un año.