MYLOTARG 5 MG POLVO PARA CONCENTRADO PARA SOLUCION PARA PERFUSION

| ATC: Gemtuzumab ozogamicina |
| PA: Gemtuzumab ozogamicina |
Envases
- Env. con 1 vial
- H: Medicamento de uso hospitalario
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica Medicamento Huérfano
Medicamento Huérfano- No sustit.: Medicamento NO sustituible por el farmacéutico (Biológicos)
- Fi: Medicamento financiado sólo para determinadas indicaciones
- Facturable SNS: NO
- Comercializado: Si
- Situación: Alta
- Código Nacional: 721601
- EAN13: 8470007216018
- Conservar en frío: Sí
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial de polvo para concentrado para solución para perfusión contiene 5 mg de gemtuzumab ozogamicina.
Tras la reconstitución (ver sección 6.6), la solución concentrada contiene 1 mg/ml de gemtuzumab ozogamicina.
Gemtuzumab ozogamicina es un conjugado de anticuerpo y fármaco (CAF) compuesto por un anticuerpo monoclonal dirigido contra CD33 (hP67.6; anticuerpo recombinante humanizado de clase inmunoglobulina [Ig]G4/kappa producido en cultivos de células de mamífero NS0) que está unido covalentemente al citotóxico N-acetil-gamma-calicheamicina.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. - FORMA FARMACÉUTICA
Polvo para concentrado para solución para perfusión (polvo para concentrado).
Pasta o polvo de color blanco a blanquecino.
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
MYLOTARG está indicado para el tratamiento combinado con daunorubicina (DNR) y citarabina (AraC) en el tratamiento de pacientes a partir de los 15 años de edad con leucemia mieloide aguda (LMA) CD-33 positiva de novo no tratada previamente, excepto la leucemia promielocítica aguda (LPA) (ver las secciones 4.4 y 5.1).
4.2 - Posología y administración de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
4.3 - Contraindicaciones de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 - Advertencias y Precauciones de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Trazabilidad
Para mejorar la trazabilidad de los medicamentos biológicos, el nombre y el número de lote del producto administrado se deben registrar claramente.
Hepatotoxicidad, que incluye enfermedad hepática venooclusiva/síndrome de obstrucción sinusoidal (EVO/SOS)
Se han notificado casos de hepatotoxicidad, incluida insuficiencia hepática potencialmente mortal y a veces mortal, y EVO/SOS en pacientes tratados con MYLOTARG (ver sección 4.8).
De acuerdo a un análisis de los posibles factores de riesgo, los pacientes adultos que recibieron MYLOTARG en monoterapia, ya sea antes o después de un trasplante de células madre hematopoyéticas (TCMH), y los pacientes con insuficiencia hepática moderada o grave tienen un mayor riesgo de presentar EVO (ver sección 4.8).
Debido al riesgo de EVO/SOS, se deben vigilar estrechamente los signos y síntomas de EVO/SOS; éstos pueden incluir elevaciones de los valores de ALT, AST, bilirrubina total y fosfatasa alcalina, que se deben controlar antes de cada dosis de MYLOTARG, hepatomegalia (que puede ser dolorosa), aumento rápido de peso y ascitis. Si se controla solo la bilirrubina total, puede que no se identifiquen todos los pacientes con riesgo de EVO/SOS. En los pacientes que presenten pruebas hepáticas anormales, se recomienda un control más frecuente de las pruebas hepáticas y de los signos y síntomas clínicos de hepatotoxicidad. En los pacientes que vayan a someterse a un TCMH, se recomienda un control estrecho de las pruebas hepáticas tras el TCMH, según proceda. No se encontró una relación definitiva entre la EVO y el tiempo de TCMH con respecto a las dosis más altas de MYLOTARG en monoterapia; sin embargo, el estudio ALFA-0701 recomendó un intervalo de 2 meses entre la última dosis de MYLOTARG y el TCMH.
El tratamiento de los signos o síntomas de toxicidad hepática puede requerir la interrupción de la dosis o suspensión del tratamiento con MYLOTARG (ver sección 4.2). En pacientes que experimenten EVO/SOS, se debe suspender el tratamiento con MYLOTARG y los pacientes deben ser tratados según la práctica médica habitual.
Reacciones asociadas a la perfusión (incluyendo anafilaxia)
En los ensayos clínicos se notificaron reacciones asociadas a la perfusión, incluyendo anafilaxia (ver sección 4.8). Se han notificado casos de reacciones asociadas a la perfusión mortales durante la fase poscomercialización. Los signos y síntomas de las reacciones asociadas a la perfusión pueden incluir fiebre y escalofríos y, con menor frecuencia, hipotensión, taquicardia y síntomas respiratorios que se pueden producir durante las primeras 24 horas tras la administración. La perfusión de MYLOTARG se debe realizar bajo estrecha vigilancia clínica, incluyendo el control del pulso, la tensión arterial y la temperatura. Se recomienda la premedicación con un corticoesteroide, antihistamínico y paracetamol 1 hora antes de la administración de MYLOTARG (ver sección 4.2). La perfusión se debe interrumpir de inmediato en pacientes que presenten indicios de reacciones graves, especialmente disnea, broncoespasmo o hipotensión clínicamente significativa. Los pacientes deben ser vigilados hasta que los signos y síntomas desaparezcan por completo. Se debe considerar seriamente la interrupción del tratamiento en los pacientes que presenten signos o síntomas de anafilaxia, incluidos los síntomas respiratorios graves o la hipotensión clínicamente significativa (ver sección 4.2).
Mielosupresión
En los ensayos clínicos, se notificaron casos de neutropenia, trombocitopenia, anemia, leucopenia, neutropenia febril, linfopenia y pancitopenia, algunos de los cuales fueron potencialmente mortales o mortales (ver sección 4.8). Las complicaciones asociadas con la neutropenia y la trombocitopenia pueden incluir infecciones y reacciones de sangrado/hemorragia, respectivamente. Se han notificado infecciones y reacciones de sangrado/hemorragia, algunos de los cuales fueron potencialmente mortales o mortales.
Se deben controlar los hemogramas completos antes de cada dosis de MYLOTARG. Durante el tratamiento, se debe vigilar a los pacientes para detectar cualquier signo y síntoma de infección, sangrado/hemorragia u otros efectos de la mielosupresión. Está indicado llevar a cabo controles rutinarios de vigilancia clínica y de laboratorio durante y después del tratamiento.
El tratamiento de pacientes con infección grave, sangrado/hemorragia u otros efectos de la mielosupresión, incluida la neutropenia grave o la trombocitopenia persistente, puede requerir un retraso de la dosis o la suspensión permanente del tratamiento con MYLOTARG (ver sección 4.2).
Síndrome de lisis tumoral (SLT)
En los ensayos clínicos se ha notificado SLT (ver sección 4.8). Se han notificado casos mortales de SLT complicados por insuficiencia renal aguda durante la fase poscomercialización. En pacientes con LMA hiperleucocitaria, se debe considerar la leucorreducción con hidroxiurea o leucoféresis para reducir el recuento de leucocitos periféricos por debajo de 30 000/mm3 antes de la administración de MYLOTARG para reducir el riesgo de inducir SLT (ver sección 4.2).
Se debe vigilar a los pacientes para detectar cualquier signo y síntoma de SLT y se deben tratar según la práctica médica habitual. Se deben tomar las medidas apropiadas para ayudar a prevenir la aparición de hiperuricemia asociada a lisis tumoral como, por ejemplo, la hidratación y la administración de antihiperuricémicos (p. ej., alopurinol) u otros fármacos para el tratamiento de la hiperuricemia (p. ej., rasburicasa).
LMA con citogenética de riesgo adverso
La eficacia de MYLOTARG se ha observado en pacientes con LMA con citogenética de riesgo favorable e intermedio, con incertidumbre respecto al tamaño del efecto en pacientes con citogenética adversa (ver sección 5.1). Cuando los resultados de las pruebas citogenéticas estén disponibles para los pacientes tratados con MYLOTARG en combinación con daunorubicina y citarabina para la LMA de novo recién diagnosticada, se debe considerar si el beneficio potencial de continuar el tratamiento con MYLOTARG supera los riesgos para cada paciente individual (ver sección 5.1).
Anticoncepción
A las mujeres en edad fértil o a las parejas de mujeres en edad fértil se les debe recomendar que utilicen 2 métodos anticonceptivos efectivos durante el tratamiento con MYLOTARG durante al menos 7 meses (mujeres) o 4 meses (hombres) después de la última dosis (ver sección 4.6).
Excipientes
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis; esto es, esencialmente “exento de sodio”.
Este medicamento puede ser preparado para la administración con soluciones que contienen sodio (ver secciones 4.2 y 6.6), y esto se debe considerar en relación con el sodio total de todas las fuentes que se administrarán al paciente.
4.5 - Interacciones con otros medicamentos de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
No se han realizado estudios de interacciones con MYLOTARG. Ver la sección 5.2 para consultar los datos disponibles de los estudios in vitro.
4.6 - Embarazo y Lactancia de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Mujeres en edad fértil/anticoncepción en hombres y mujeres
A las mujeres en edad fértil se les debe recomendar que eviten quedarse embarazadas durante el tratamiento con MYLOTARG.
A las mujeres en edad fértil o las parejas de mujeres en edad fértil se les debe recomendar que utilicen 2 métodos anticonceptivos efectivos durante el tratamiento con MYLOTARG durante al menos 7 meses (mujeres) o 4 meses (hombres) después de la última dosis.
Embarazo
No hay datos o éstos son limitados relativos al uso de gemtuzumab ozogamicina en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
MYLOTARG no se debe utilizar durante el embarazo a menos que el beneficio potencial para la madre supere los riesgos potenciales para el feto. Se debe informar del riesgo potencial para el feto a las mujeres embarazadas, a las pacientes que se queden embarazadas mientras reciben gemtuzumab ozogamicina, o a los varones tratados cuya pareja esté embarazada.
Lactancia
No se dispone de información relativa a la presencia de gemtuzumab ozogamicina o sus metabolitos en la leche materna, los efectos sobre el lactante o los efectos sobre la producción de leche. Debido a la posibilidad de reacciones adversas debidas al medicamento en los lactantes, las mujeres no deben dar el pecho durante el tratamiento con MYLOTARG y hasta al menos 1 mes después de la última dosis (ver sección 5.3).
Fertilidad
No se dispone de información relativa a la fertilidad en los pacientes. Según los hallazgos preclínicos, la fertilidad masculina y femenina se puede ver comprometida con el tratamiento con gemtuzumab ozogamicina (ver sección 5.3). Tanto los hombres como las mujeres deben solicitar información sobre la preservación de su fertilidad antes del tratamiento.
4.7 - Efectos sobre la capacidad de conducción de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
La influencia de MYLOTARG sobre la capacidad para conducir y utilizar máquinas es moderada. Se debe informar a los pacientes de que pueden experimentar fatiga, mareos y cefalea durante el tratamiento con MYLOTARG (ver sección 4.8). Por lo tanto, se debe tener precaución al conducir o utilizar máquinas.
4.8 - Reacciones Adversas de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Resumen del perfil de seguridad
El perfil de seguridad general de MYLOTARG se basa en los datos de pacientes con leucemia mieloide aguda obtenidos del estudio de tratamiento combinado ALFA-0701, de los estudios en monoterapia y de la experiencia poscomercialización. En el estudio de terapia combinada, los datos de seguridad compuestos por las reacciones adversas surgidas durante el tratamiento (TEAE, por sus siglas en inglés) seleccionadas que se consideraban esenciales para comprender el perfil de seguridad de MYLOTARG consistieron en hemorragias de todos los grados, EVO de todos los grados e infecciones graves. Todas estas TEAE se determinaron como reacciones adversas debidas al medicamento. Debido a esta limitada recopilación de datos, los datos de laboratorio del estudio de tratamiento combinado se incluyen en la tabla 5. La información sobre las reacciones adversas debidas al medicamento procedentes de los estudios en monoterapia que utilizan la pauta no fraccionada (estudios 201/202/203) y de la experiencia poscomercialización se presenta en la tabla 6, mientras que el estudio en monoterapia B1761031 que utiliza la pauta fraccionada se presenta en la sección siguiente, para proporcionar una caracterización completa de las reacciones adversas debidas al medicamento.
En el estudio de tratamiento combinado ALFA-0701, las reacciones adversas graves debidas al medicamento clínicamente importantes fueron hepatotoxicidad, incluyendo EVO/SOS (3,8 %), hemorragia (9,9 %), infección grave (41,2 %) y síndrome de lisis tumoral (1,5 %). En los estudios en monoterapia (estudios 201/202/203), las reacciones adversas graves clínicamente relevantes debidas al medicamento también incluyeron reacciones relacionadas con la perfusión (2,5 %), trombocitopenia (21,7 %) y neutropenia (34,3 %). En el estudio en monoterapia B1761031, las reacciones adversas graves clínicamente relevantes debidas al medicamento, incluyeron infección (30,0 %), neutropenia febril (22,0 %), pirexia (6,0 %), hemorragia (4,0 %), trombocitopenia (4,0 %), anemia (2,0 %) y taquicardia (2,0 %).
Las reacciones adversas debidas al medicamento más frecuentes (> 30 %) en el estudio de terapia combinada fueron hemorragia e infección. En los estudios en monoterapia (estudios 201/202/203), las reacciones adversas debidas al medicamento más frecuentes (> 30 %) incluyeron pirexia, náusea, infección, escalofríos, hemorragia, vómitos, trombocitopenia, fatiga, cefalea, estomatitis, diarrea, dolor abdominal y neutropenia. En el estudio en monoterapia B1761031, las reacciones adversas debidas al medicamento más frecuentes (> 30 %) incluyeron infección (50,0 %), neutropenia febril (40,0 %) y hemorragia (32,0 %).
Las reacciones adversas debidas al medicamento más frecuentes (≥ 1 %) que llevaron a la interrupción permanente en el estudio de tratamiento combinado fueron trombocitopenia, EVO, hemorragia e infección. Las reacciones adversas debidas al medicamento más frecuentes (≥ 1 %) que llevaron a la interrupción permanente en los estudios en monoterapia (estudios 201/202/203) fueron infección, hemorragia, insuficiencia multiorgánica y EVO. Las reacciones adversas debidas al medicamento que llevaron a la interrupción permanente en el estudio en monoterapia B1761031 fueron infección y pirexia.
Tabla de reacciones adversas debidas al medicamento
Las reacciones adversas debidas al medicamento se presentan conforme a la clasificación por órganos y sistemas (SOC, por sus siglas en inglés) y por categorías de frecuencia, definidas mediante la siguiente convención: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1 000 a < 1/100), raras (≥ 1/10 000 a < 1/1 000), muy raras (< 1/10 000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las reacciones adversas debidas al medicamento se presentan en orden decreciente de gravedad.
Tabla 5. Reacciones adversas debidas al medicamento seleccionadas** en pacientes que recibieron MYLOTARG en el estudio de tratamiento combinado (ALFA-0701)
| Daunorubicina + citarabina (N = 137) | ||||
| Todos los grados % | Todos los grados % | |||
| Enfermedad hepática venooclusiva*c | 2,3 | 1,5 | ||
| Exploraciones complementarias *** | ||||
| Muy frecuentes | ||||
| Hemoglobina disminuida | 86,2 | 89,7 | ||
| Plaquetas disminuidas | 100 | 100 | ||
| Leucocitos disminuidos | 100 | 99,3 | ||
| Linfocitos (absolutos) disminuidos | 90,7 | 89,6 | ||
| Neutrófilos disminuidos | 96,1 | 97,0 | ||
| Hiperglucemia | 19,2 | 17,8 | ||
| Aspartato aminotransferasa (AST) elevada | 14,0 | 9,0 | ||
| Tiempo de protrombina prolongado | 3,3 | 0 | ||
| Tiempo de tromboplastina parcial activada prolongado | 6,4 | 5,5 | ||
| Fosfatasa alcalina elevada | 13,3 | 5,3 | ||
| Alanina aminotransferasa (ALT) elevada | 10,9 | 15,7 | ||
| Bilirrubina en sangre elevada | 7,1 | 3,8 | ||
| Hiperuricemia | 2,6 | 0 | ||
Abreviaturas: N = número de pacientes; TP = término preferente.
*Con desenlace mortal.
**Solo se recopilaron datos de seguridad seleccionados en este estudio de LMA recién diagnosticada.
***Frecuencia basada en valores de laboratorio (grado según el Instituto Nacional del Cáncer de los EEUU [CTCAE del NCI] v4.03).
a. Infección incluye sepsis y bacteriemia (53,4 %), infección fúngica (15,3 %), infección del tracto respiratorio inferior (5,3 %), infección bacteriana (9,2 %), infección gastrointestinal (8,4 %), infección cutánea (2,3 %) y otras infecciones (28,4 %).
b. Hemorragia incluye hemorragia del sistema nervioso central (3,1 %), hemorragia gastrointestinal alta (33,6 %), hemorragia gastrointestinal baja (17,6 %), hemorragia subcutánea (60,3 %), otras hemorragias (64,9 %) y epistaxis (62,6 %).
c. Enfermedad hepática venooclusiva incluye los siguientes términos preferentes notificados: enfermedad venooclusiva y enfermedad hepática venooclusiva*.
Tabla 6. Reacciones adversas debidas al medicamento en pacientes que recibieron MYLOTARG en los estudios en monoterapia*** y durante la experiencia poscomercialización
| Clasificación por órganos y sistemas Frecuencia Término preferente | Grado 3/4 % |
| Fosfatasa alcalina en sangre elevada | 6,1 |
*Incluye desenlace mortal.
**Incluye reacciones adversas debidas al medicamento mortales en la fase poscomercialización.
***MYLOTARG en el tratamiento de la recaída de LMA (9 mg/m2) (estudios 201/202/203).
#Casos individuales.
Abreviatura: TP = término preferente.
a. Infección incluye sepsis y bacteriemia (25,6 %), infección fúngica (10,5 %), infección del tracto respiratorio inferior (13,0 %), infección del tracto respiratorio superior (4,3 %), infección bacteriana (3,6 %), infección vírica (24,2 %), infección gastrointestinal (3,3 %), infección cutánea (7,9 %) y otras infecciones (19,5 %). También se notificaron infecciones pulmonares fúngicas posteriores a la comercialización (categoría de frecuencia no conocida) que incluyen micosis pulmonar y neumonía por Pneumocystis jirovecii*; e infecciones bacterianas, incluida la infección por Stenotrophomonas.
b. Trombocitopenia incluye los siguientes TP notificados: recuento de plaquetas disminuido y trombocitopenia*.
c. Neutropenia incluye los siguientes TP notificados: neutropenia, granulocitopenia y recuento de neutrófilos disminuido.
d. Anemia incluye los siguientes TP notificados: anemia y hemoglobina disminuida.
e. Leucopenia incluye los siguientes TP notificados: leucopenia y recuento de leucocitos disminuido.
f. Pancitopenia incluye los siguientes TP notificados: pancitopenia e insuficiencia de médula ósea.
g. Linfopenia incluye los siguientes TP notificados: linfopenia y recuento de linfocitos disminuido.
h. Reacción asociada a la perfusión incluye los siguientes TP notificados: reacción asociada a la perfusión, urticaria, hipersensibilidad, broncoespasmo, hipersensibilidad al medicamento y urticaria en la zona de inyección#.
i. Hiperglucemia incluye los siguientes TP notificados: hiperglucemia y glucosa en sangre elevada#.
j. Taquicardia incluye los siguientes TP notificados: taquicardia, taquicardia sinusal, frecuencia cardiaca aumentada# y taquicardia supraventricular#.
k. Hemorragia incluye hemorragia del sistema nervioso central (5,1 %), hemorragia gastrointestinal alta (21,3 %), hemorragia gastrointestinal baja (15,2 %), hemorragia subcutánea (28,5 %), otras hemorragias (32,9 %) y epistaxis (28,5 %).
l. Hipotensión incluye los siguientes TP notificados: hipotensión y tensión arterial disminuida.
m. Hipertensión incluye los siguientes TP notificados: hipertensión y tensión arterial aumentada.
n. Disnea incluye los siguientes TP notificados: disnea y disnea de esfuerzo.
o. Dolor abdominal incluye los siguientes TP notificados: dolor abdominal, dolor abdominal inferior, dolor abdominal superior, molestia abdominal y dolor a la palpación abdominal.
p. Estomatitis incluye los siguientes TP notificados: inflamación de mucosa, dolor orofaríngeo, estomatitis, ulceración de boca, dolor bucal, ampollas en la mucosa bucal, estomatitis aftosa, ulceración de la lengua, glosodinia, eritema de mucosa bucal, glositis# y ampollas orofaríngeas#.
q. Transaminasas elevadas incluye los siguientes TP notificados: transaminasas elevadas, lesión hepatocelular, alanina aminotransferasa elevada, aspartato aminotransferasa elevada y enzimas hepáticas aumentadas.
r.Hiperbilirrubinemia incluye los siguientes TP notificados: bilirrubina en sangre elevada e hiperbilirrubinemia.
s. Enfermedad hepática venooclusiva incluye los siguientes TP notificados: enfermedad venooclusiva y enfermedad hepática venooclusiva*#.
t. Función hepática anormal incluye los siguientes TP notificados: prueba de función hepática anormal y función hepática anormal.
u. Erupción incluye los siguientes TP notificados: erupción, dermatitis#, dermatitis alérgica#, dermatitis ampollosa, dermatitis de contacto, dermatitis exfoliativa#, erupción medicamentosa, prurito alérgico# y erupción eritematosa#, erupción macular#, erupción maculopapular, erupción papular, erupción pruriginosa, erupción vesicular#.
v. Eritema incluye los siguientes TP notificados: eritema en el lugar de entrada de un catéter, eritema y eritema en la zona de perfusión#.
w. Pirexia incluye los siguientes TP notificados: pirexia, temperatura corporal elevada e hipertermia.
x. Edema incluye los siguientes TP notificados: Edema, edema facial, edema periférico, hinchazón de cara, edema generalizado y edema periorbital.
y. Fatiga incluye los siguientes TP notificados: fatiga, astenia, letargo y malestar.
Descripción de reacciones adversas seleccionadas
Hepatotoxicidad, incluida EVO/SOS hepático
En el estudio de tratamiento combinado, se recopilaron las analíticas hepáticas con alteraciones y la EVO. Los estudios en monoterapia proporcionan una caracterización adicional de las reacciones adversas hepatotóxicas.
En el estudio de terapia combinada (N = 131), se notificó EVO en 6 (4,6%) pacientes durante o después del tratamiento, 2 (1,5%) de estas reacciones fueron mortales (ver tabla 5). Cinco (3,8%) de estas reacciones de EVO se produjeron en los 28 días siguientes a la administración de cualquier dosis de gemtuzumab ozogamicina. Un caso de EVO se produjo después de 28 días tras la última dosis de gemtuzumab ozogamicina y otro de esos casos se produjo unos días después de haber comenzado una pauta de acondicionamiento para un TCMH. La mediana del tiempo desde la última dosis de gemtuzumab ozogamicina hasta la aparición de EVO fue de 9 días (intervalo: 2 - 298 días). También se notificó EVO en 2 pacientes que recibieron MYLOTARG como tratamiento de seguimiento después de una recaída de LMA tras el tratamiento con quimioterapia en el grupo control del estudio de tratamiento combinado. Ambos pacientes experimentaron EVO después de 28 días tras la última dosis de gemtuzumab ozogamicina. Uno de estos pacientes experimentó EVO 25 días después del TCMH posterior.
En el estudio en monoterapia B1761031, no se notificaron acontecimientos de EVO en ningún paciente. Sin embargo, 1 (2,0 %) paciente presentó síndrome de fuga capilar mortal con síntomas concordantes con EVO (ascitis e hiperbilirrubinemia). Los acontecimientos de hepatotoxicidad grado 3 incluyeron aumento de gamma-glutamiltransferasa (4,0 %), aumento de alanina aminotransferasa (2,0 %), aumento de aspartato aminotransferasa (2,0 %), hipoalbuminemia (2,0 %) y aumento de transaminasas (2,0 %). Ningún paciente presentó hepatotoxicidad de grado 4 o grado 5.
Según un análisis de los posibles factores de riesgo sobre los pacientes adultos que recibieron MYLOTARG no fraccionado en monoterapia, se observó que los pacientes que se sometieron a un TCMH antes de la exposición a gemtuzumab ozogamicina fueron 2,6 veces más propensos (intervalo de confianza [IC] del 95%: 1,448; 4,769) a presentar EVO que los pacientes sin TCMH antes del tratamiento con gemtuzumab ozogamicina; los pacientes que habían recibido un TCMH tras el tratamiento con gemtuzumab ozogamicina fueron 2,9 veces más propensos (IC del 95%: 1,502; 5,636) a presentar EVO que los pacientes sin TCMH tras el tratamiento con gemtuzumab ozogamicina; y los pacientes que padecían insuficiencia hepática moderada/grave al inicio del estudio fueron 8,7 veces más propensos (IC del 95%: 1,879; 39,862) a presentar EVO que los pacientes que no padecían insuficiencia hepática moderada/grave al inicio del estudio.
Los pacientes deben ser vigilados por si presentan hepatotoxicidad como se recomienda en sección 4.4. El tratamiento de los signos o síntomas de toxicidad hepática puede requerir la interrupción de la dosis o la suspensión del tratamiento con MYLOTARG (ver sección 4.2).
Mielosupresión
En el estudio de terapia combinada en pacientes con LMA de novo no tratada y tratados previamente con dosis fraccionadas de gemtuzumab ozogamicina en combinación con quimioterapia, se observaron disminuciones de grado 3/4 de leucocitos, neutrófilos y plaquetas en 131 (100%), 124 (96,1%) y 131 (100%) pacientes, respectivamente.
Durante la fase de inducción, 109 (83,2%) y 99 (75,6%) pacientes presentaron recuperación del recuento plaquetario a 50 000/mm3 y 100 000/mm3, respectivamente. La mediana de tiempo hasta la recuperación del recuento plaquetario a 50 000/mm3 y 100 000/mm3 fue de 34 y 35 días, respectivamente. Durante la fase de consolidación 1, 92 (94,8%) y 71 (73,2%) pacientes presentaron recuperación del recuento plaquetario a 50 000/mm3 y 100 000/mm3, respectivamente. La mediana de tiempo hasta la recuperación del recuento plaquetario a 50 000/mm3 y 100 000/mm3 fue de 32 y 35 días, respectivamente. Durante la fase de consolidación 2, 80 (97,6%) y 70 (85,4%) pacientes presentaron recuperación del recuento plaquetario a 50 000/mm3 y 100 000/mm3, respectivamente. La mediana de tiempo hasta la recuperación del recuento plaquetario a 50 000/mm3 y 100 000/mm3 fue de 36,5 y 43 días, respectivamente.
Se produjo trombocitopenia con recuentos plaquetarios <50 000/mm3 que persistieron durante 45 días tras el inicio del tratamiento para los pacientes que respondieron (RC y recuperación plaquetaria incompleta [RCp]) en 22 (20,4%) de los pacientes. El número de pacientes con trombocitopenia persistente se mantuvo más o menos igual en todos los ciclos de tratamiento (8 [7,4%] pacientes en la fase de inducción, 8 [8,5%] pacientes en la fase de consolidación 1 y 10 [13,2%] pacientes en la fase de consolidación 2).
Durante la fase de inducción, 121 (92,4%) y 118 (90,1%) pacientes presentaron recuperación de neutrófilos documentada a un RAN de 500/mm3 y 1 000/mm3, respectivamente. La mediana de tiempo hasta la recuperación de neutrófilos a un RAN de 500/mm3 y 1 000/mm3 fue de 25 días. Durante la fase de consolidación 1 del tratamiento, 94 (96,9%) pacientes presentaron recuperación de neutrófilos a recuentos de 500/mm3 y 91 (94%) pacientes presentaron recuperación a recuentos de 1 000/mm3. La mediana de tiempo hasta la recuperación de neutrófilos a un RAN de 500/mm3 y 1 000/mm3 fue de 21 y 25 días, respectivamente. Durante la fase de consolidación 2 de la terapia, 80 (97,6%) pacientes presentaron recuperación de neutrófilos a recuentos de 500/mm3 y 79 (96,3%) pacientes presentaron recuperación a recuentos de 1 000/mm3. La mediana de tiempo hasta la recuperación de neutrófilos a un RAN de 500/mm3 y 1 000/mm3 fue de 22 y 27 días, respectivamente.
En el estudio de terapia combinada en pacientes con LMA de novo tratados con dosis fraccionadas de gemtuzumab ozogamicina en combinación con quimioterapia (N = 131), 102 (77,9%) pacientes experimentaron infecciones graves por cualquier causa (grado ≥ 3). Se notificó muerte relacionada con el tratamiento por shock séptico en 1 (0,8%) paciente. Se notificó infección grave mortal en 2 (1,53%) pacientes del grupo de MYLOTARG y en 4 (2,92%) pacientes del grupo control.
En el estudio de tratamiento combinado (N = 131), se notificaron reacciones de sangrado/hemorragia de todos los grados y de grado 3/4 en 118 (90,1%) y 27 (20,6%) pacientes, respectivamente. Las reacciones de sangrado/hemorragia de grado 3 más frecuentes fueron hematemesis (3,1%), hemoptisis (3,1%) y hematuria (2,3%). Se notificaron reacciones de sangrado/hemorragia de grado 4 en 4 (3,1%) pacientes (hemorragia gastrointestinal, hemorragia y hemorragia alveolar pulmonar [2 pacientes]). Se notificaron reacciones de sangrado/hemorragia mortales en 3 (2,3%) pacientes (hematoma cerebral, hematoma intracraneal y hematoma subdural).
En el estudio en monoterapia B1761031 (N = 50), se notificaron infecciones de grado 3/4 en 10 (20%) pacientes. Las infecciones de grado 3/4 notificadas con mayor frecuencia (≥ 5,0%) fueron sepsis y neumonía en 3 (6,0%) pacientes cada una de ellas. Seis (6) (12,0%) pacientes presentaron infección de grado 5 (sepsis en 4 [8,0%] pacientes, y neumonía atípica y neumonía por COVID-19 en 1 [2,0%] paciente cada una). Se notificaron acontecimientos de sangrado/hemorragia de cualquier grado en 16 (32,0%) pacientes. Se produjeron acontecimientos de hemorragia de grado 3/4 en 2 (4,0%) pacientes (hemorragia gástrica de grado 3 y hemorragia intracraneal traumática de grado 4 en 1 paciente cada una). No se notificaron acontecimientos mortales de sangrado/hemorragia.
El tratamiento de pacientes con infección grave, sangrado/hemorragia u otros efectos de la mielosupresión, incluida la neutropenia grave o la trombocitopenia persistente, puede requerir un retraso de la dosis o la suspensión permanente del tratamiento con MYLOTARG (ver las secciones 4.2 y 4.4).
Inmunogenicidad
Al igual que ocurre con todas las proteínas terapéuticas, existe potencial de inmunogenicidad.
En el estudio en monoterapia B1761031 en 50 pacientes adultos con LMA CD33 positiva recidivante o refractaria, se evaluó el anticuerpo frente al medicamento (ADA, por sus siglas en inglés) frente a MYLOTARG mediante el método de electroquimioluminiscencia (EQL). Para los pacientes cuyas muestras de ADA dieron positivo, se desarrolló un análisis en células aisladas para medir los anticuerpos neutralizantes (NAb, por sus siglas en inglés) frente a MYLOTARG.
La incidencia de ADA y NAb fue de 6 (12,0%) y 1 (2,0%), respectivamente. La presencia de ADA no tuvo efectos estadísticamente significativos o clínicamente relevantes sobre la FC del anticuerpo hP67.6 total o la calicheamicina conjugada. Ninguno de los pacientes experimentó anafilaxia, hipersensibilidad u otras secuelas clínicas relacionadas con el ADA. No hubo indicios de que la presencia de ADA tuviera una relación directa con posibles problemas de seguridad.
La detección de ADAs depende en gran medida de la sensibilidad y especificidad del ensayo. La incidencia de resultados positivos en la prueba de anticuerpos se puede ver influenciada por varios factores, incluida la metodología de la prueba, las concentraciones de gemtuzumab ozogamicina circulante, el manejo de las muestras, el momento de la toma de la muestra, los tratamientos concomitantes y la enfermedad subyacente. Por estas razones, comparar la incidencia de anticuerpos frente a gemtuzumab ozogamicina con la incidencia de anticuerpos frente a otros medicamentos puede inducir a error.
Población pediátrica
LMA no tratada previamente
No se ha establecido la seguridad y eficacia de MYLOTARG en niños y adolescentes menores de 15 años con LMA no tratada previamente (ver sección 4.2).
En el estudio pediátrico completado de fase 3 aleatorizado AAML0531 (ver sección 5.1) de gemtuzumab ozogamicina combinado con tratamiento intensivo de primera línea en 1 063 niños recién diagnosticados (93,7% de los pacientes < 18 años de edad) y adultos jóvenes (6,3% de los pacientes) con LMA de novo de 0 a 29 años, el perfil de seguridad fue similar al observado en los otros estudios de gemtuzumab ozogamicina combinado con quimioterapia intensiva en pacientes adultos con LMA de novo. Sin embargo, no se estableció ninguna dosis óptima de gemtuzumab ozogamicina para pacientes pediátricos, ya que durante el segundo periodo de intensificación tras la segunda dosis de gentuzumab ozogamicina del estudio AAML0531, una mayor proporción de pacientes del grupo de gemtuzumab ozogamicina experimentó un tiempo de recuperación de neutrófilos prolongado (> 59 días) comparado con el grupo comparador (21,0% frente a 11,5%), y más pacientes murieron durante la remisión (5,5% frente a 2,8%).
LMA recidivante o refractaria
No se ha establecido la seguridad y eficacia de MYLOTARG en pacientes pediátricos con LMA recidivante o refractaria (ver secciones 4.1 y 4.2).
Los resultados de seguridad observados en una revisión sistemática de la literatura médica de estudios que han evaluado MYLOTARG en pacientes pediátricos (ver sección 5.1) se presentan en la tabla 7.
Tabla 7. Resultados de seguridad de una revisión sistemática de la literatura médica en pacientes pediátricos con LMA recidivante o refractaria que recibieron MYLOTARG
| Monoterapia | Combinacióna | |||||||||||
| MYLOTARG fraccionadob | MYLOTARG no fraccionadob | MYLOTARG fraccionadob | MYLOTARG no fraccionadob | |||||||||
| Número de estudios | N por estudio (intervalo) | Tasac (%) | Número de estudios | N por estudio (intervalo) | Tasac (%) | Número de estudios | N por estudio (intervalo) | Tasac (%) | Número de estudios | N por estudio (intervalo) | Tasac (%) | |
| EVO | 1 | 6 | 0 | 10 | 5, 30 | 6,8 | 2 | 3, 17 | 0 | 5 | 5, 84 | 4,4 |
| EVO tras el TCMH | No notificada | 5 | 4, 14 | 19,1 | 2 | 3, 8 | 0 | 2 | 12, 28 | 14,7 | ||
| Muerted | 1 | 6 | 0 | 4 | 6, 29 | 10,8 | No notificada | 3 | 5, 45 | 6,5 | ||
| Infección | 5 estudios; N por estudio (intervalo) 12-30; 28,4% | 4 estudios; N por estudio (intervalo) 12-84; 42,2% | ||||||||||
| Mielosupresióne | Casi todos los pacientes (> 90%) experimentaron mielosupresión en todos los estudios | |||||||||||
| a: Cuando MYLOTARG se administró en combinación, citarabina fue parte de la combinación estudiada en 8 de los 9 estudios. b: Dosis fraccionadas hace referencia a una pauta posológica de MYLOTARG de 3 mg/m2 en los días 1, 4 y 7. Dosis no fraccionadas hace referencia a la administración de MYLOTARG (intervalo de dosis total 1,8 mg/m2–9 mg/m2) 2 veces durante un ciclo con al menos 14 días de diferencia. c: Las tasas en todos los estudios se calcularon mediante la ponderación de la varianza inversa con efectos fijos. Las proporciones se transformaron utilizando la transformación arcoseno doble de Freeman-Tukey antes de combinar los estudios, y la tasa combinada estimada se transformó nuevamente utilizando la media armónica de los tamaños de las muestras del estudio. d: En los 30 días posteriores a la última dosis de MYLOTARG. e: Cuando se analizó, la mediana de la recuperación (definida como 20 x 109/l o 50 x 109/l para plaquetas y 0,5 x 109/l para neutrófilos) osciló entre 42-48 días para plaquetas y 30-37 días para neutrófilos. | ||||||||||||
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es
4.9 - Sobredosificación de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
No se han notificado casos de sobredosis con MYLOTARG durante la experiencia clínica. No se han probado dosis únicas superiores a 9 mg/m2 en adultos. El tratamiento de la sobredosis de MYLOTARG debe consistir en medidas de soporte generales.
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Grupo farmacoterapéutico: agentes antineoplásicos, anticuerpos monoclonales y anticuerpos conjugados a fármaco, otros anticuerpos monoclonales y anticuerpos conjugados a fármaco, código ATC: L01FX02
Mecanismo de acción
Gemtuzumab ozogamicina es un CAF dirigido contra CD33. Gemtuzumab es un anticuerpo humanizado de inmunoglobulina de clase G subtipo 4 (IgG4) que reconoce específicamente CD33 humano. La porción de anticuerpo se une específicamente al antígeno CD33, una proteína de adhesión dependiente de ácido siálico que se encuentra en la superficie de los linfoblastos leucémicos mieloides y de las células normales inmaduras de linaje mielomonocítico, pero no en las células madre hematopoyéticas normales. La molécula pequeña, N-acetil-gamma-calicheamicina, es un producto natural semisintético citotóxico. N-acetil-gamma-calicheamicina se une de forma covalente al anticuerpo mediante un enlazador AcBut [ácido 4-(4-acetilfenoxi) butanoico]. Los datos preclínicos sugieren que la actividad anticancerosa de gemtuzumab ozogamicina se debe a la unión del CAF a las células cancerígenas que expresan CD33, seguido por la internalización del complejo CAF-CD33 y la liberación intracelular de N-acetil-gamma-calicheamicina dimetilhidrazida mediante la hidrólisis del enlazador. La activación de N-acetil-gamma-calicheamicina dimetilhidrazida induce roturas del ADN bicatenario, provocando posteriormente la interrupción del ciclo celular y la muerte celular apoptótica.
Se supone que es necesaria la saturación de un alto porcentaje de sitios antigénicos de CD33 para la distribución máxima de calicheamicina a los linfoblastos leucémicos. Varios estudios en monoterapia midieron la dosis de saturación de CD33 tras la administración de MYLOTARG en pacientes con LMA recidivante y refractaria. En todos los estudios, se observó una saturación de CD33 periférica casi máxima tras la administración de MYLOTARG en todos los niveles de dosis de 2 mg/m2 y superiores, lo que sugiere que una dosis baja de gemtuzumab ozogamicina es suficiente para cubrir todos los sitios de CD33 disponibles.
Eficacia clínica y seguridad
Estudio ALFA-0701 con pacientes con LMA de novo no tratados previamente
La eficacia y la seguridad de MYLOTARG se evaluaron en un estudio de fase 3, multicéntrico, aleatorizado y abierto que comparó la incorporación de MYLOTARG a una pauta de inducción de quimioterapia habitual con la combinación daunorubicina y citarabina (combinación DA) frente a la DA sola. Los pacientes elegibles tenían entre 50 y 70 años de edad con LMA de novo previamente no tratada (estudio ALFA-0701). Los pacientes con leucemia promielocítica aguda (LPA, LMA3) y los pacientes con LMA procedente de un síndrome mielodisplásico (SMD) o LMA secundaria fueron excluidos del estudio.
La variable primaria fue la supervivencia libre de eventos (SLE). Las variables secundarias incluyeron las tasas de RC y RCp, la supervivencia libre de recaída (SLR), la supervivencia global (SG) y la seguridad de la combinación DA con o sin MYLOTARG.
En total, 271 pacientes fueron aleatorizados en este estudio con 135 pacientes asignados al tratamiento de inducción de 3 + 7 DA más dosis fraccionadas de 3 mg/m2 × 3 dosis de MYLOTARG y 136 pacientes asignados a 3 + 7 DA solamente (ver sección 4.2). Se permitió un segundo ciclo de tratamiento de inducción con DA pero sin MYLOTARG, con independencia del grupo de aleatorización. Los pacientes de cualquiera de los grupos que no recibieron el segundo ciclo de tratamiento de inducción y no lograron una RC después de la inducción pudieron recibir un ciclo de rescate compuesto de idarubicina, AraC y factor estimulante de colonias de granulocitos (G-CSF, por sus siglas en inglés).
Los pacientes con RC o RCp recibieron tratamiento de consolidación con 2 ciclos de tratamiento que incluyeron DNR y AraC con o sin MYLOTARG de acuerdo con su aleatorización inicial. Los pacientes que experimentaron remisión también fueron elegibles para el trasplante alogénico. Se recomendó un intervalo de al menos 2 meses entre la última dosis de MYLOTARG y el trasplante.
En general, la mediana de edad de los pacientes fue de 62 años (intervalo de 50 a 70 años) y la mayoría de los pacientes (87,8%) presentaban un estado funcional del Grupo Oncológico Cooperativo del Este (ECOG, por sus siglas en inglés) de 0 a 1 al inicio del estudio. Las características iniciales se equilibraron entre los grupos de tratamiento, con la excepción del sexo, ya que se reclutó a un mayor porcentaje de hombres en el grupo de MYLOTARG (54,8%) que en el grupo de DA solo (44,1%). En general el 59,0% y el 65,3% de los pacientes padecían una enfermedad documentada de riesgo favorable/intermedio según las clasificaciones del riesgo de la Red Nacional de Centros Oncológicos Integrales de EEUU (NCCN, por sus siglas en inglés) y de la Red Europea de Leucemia (ELN, por sus siglas en inglés) de 2010, respectivamente. Se determinó la expresión de CD33 en linfoblastos de LMA mediante citometría de flujo armonizada a partir de los resultados de laboratorios locales en 194/271 (71,6%) pacientes en total. Pocos pacientes (13,7%) tenían baja expresión de CD33 (menos del 30% de los linfoblastos).
El estudio cumplió su objetivo principal de demostrar que MYLOTARG añadido en dosis fraccionadas (3 mg/m2 × 3 dosis) a la quimioterapia de inducción estándar para pacientes con LMA de novo no tratada previamente conlleva a una mejoría estadísticamente significativa y clínicamente satisfactoria de la SLE. La mediana de la SLE fue de 17,3 meses (IC del 95%: 13,4; 30,0) en el grupo de MYLOTARG frente a 9,5 meses (IC del 95%: 8,1; 12,0) en el grupo de DA solo; hazard ratio (HR) 0,562 (IC del 95%: 0,415; 0,762); valor p bilateral = 0,0002 según el test de log-rank. Los datos de eficacia del estudio ALFA-0701 se resumen en la tabla 8, y la curva de Kaplan-Meier de la SLE se muestra en la figura 1.
Tabla 8. Datos de eficacia del estudio ALFA-0701 (población por IDTM)
| MYLOTARG + daunorubicina + citarabina | Daunorubicina + citarabina | |
| Supervivencia libre de eventos (por el investigador) | N = 135 | N = 136 |
| Número de eventos, n (%) | 73 (54,1) | 102 (75,0) |
| Mediana de la SLE en meses [IC del 95%]a | 17,3 [13,4-30,0] | 9,5 [8,1-12,0] |
| Probabilidad de SLE a 2 años [IC del 95%]b | 42,1 [32,9-51,0] | 18,2 [11,1-26,7] |
| Probabilidad de SLE a 3 años [IC del 95%]b | 39,8 [30,2-49,3] | 13,6 [5,8-24,8] |
| Hazard Ratio [IC del 95%]c | 0,562 [0,415-0,762] | |
| Valor pd | 0,0002 | |
| Supervivencia libre de recaída (por el investigador) | N = 110 | N = 100 |
| Número de eventos, n (%) | 49 (44,5) | 66 (66,0) |
| Mediana de la SLR en meses [IC del 95%]a | 28,0 [16,3-NE] | 11,4 [10,0-14,4] |
| Hazard Ratio [IC del 95%]c | 0,526 [0,362-0,764] | |
| Valor pd | 0,0006 | |
| Supervivencia global | N = 135 | N = 136 |
| Número de muertes, n (%) | 80 (59,3) | 88 (64,7) |
| Mediana de la SG en meses [IC del 95%]a | 27,5 [21,4-45,6] | 21,8 [15,5-27,4] |
| Hazard Ratio [IC del 95%]c | 0,807 [0,596-1,093] | |
| Valor pd | 0,1646 | |
| Tasa de respuesta (por el investigador) | N = 135 | N = 136 |
| Respuesta global % [IC del 95%]e | 81,5 [73,89-87,64] | 73,5 [65,28-80,72] |
| RC | 70,4 | 69,9 |
| RCp | 11,1 | 3,7 |
| Valor pg | 0,1457 |
Según la definición principal de SLE: fechas de los eventos (fracaso de inducción, recaída o muerte) determinadas por la evaluación del investigador.
La población por IDTM incluyó a todos los pacientes aleatorizados, a menos que se retirara el consentimiento antes del inicio del tratamiento y se analizaron de acuerdo con el grupo de aleatorización inicial.
Abreviaturas: RC = remisión completa; RCp = remisión completa con recuperación plaquetaria incompleta; IC = intervalo de confianza; SLE = supervivencia libre de eventos; IDTM = intención de tratar modificada; n = número; N = número; NE = no estimable; SG = supervivencia global; SLR = supervivencia libre de recaída.
a. Mediana estimada usando el método Kaplan-Meier; IC basado en el método Brookmeyer-Crowley con transformación logarítmica en los dos ejes.
b. Estimada a partir de la curva Kaplan-Meier. Probabilidad (%) calculada por el método del límite de producto; IC calculado a partir de la transformación logarítmica en los dos ejes de la probabilidad de supervivencia utilizando una aproximación normal y la fórmula de Greenwood.
c. Según el modelo de riesgos proporcionales de Cox frente a daunorubicina + citarabina.
d. Valor p bilateral del test de log-rank.
e. Respuesta definida como RC + RCp.
f. Diferencia de respuesta global; IC según el método de Santner y Snell.
g. Según la prueba exacta de Fisher.
Figura 1. Curva Kaplan-Meier de la supervivencia libre de eventos según la evaluación del investigador del estudio ALFA 0701 (población por IDTM)
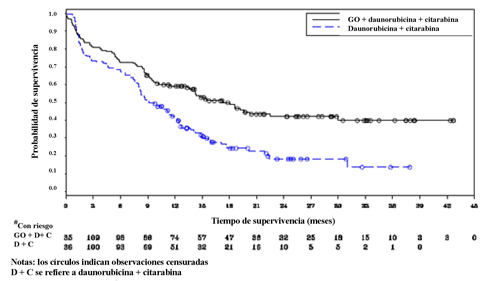
Abreviaturas: C = citarabina; D = daunorubicina; GO = gemtuzumab ozogamicina; IDTM = intención de tratar modificada.
Uso en LMA con citogenética de riesgo adverso
En los análisis de subgrupos en ALFA-0701, la incorporación de MYLOTARG a la quimioterapia combinada estándar no mejoró la SLE en el subgrupo de pacientes con citogenética de riesgo adverso (HR 1,11; IC del 95%: 0,63; 1,95). La SLE y SG analizadas conforme a la clasificación de riesgo citogenético y a la clasificación de riesgo citogenético/molecular se presentan a continuación en la tabla 9 y en la tabla 10.
Tabla 9. Supervivencia libre de eventos evaluada por el investigador conforme a las clasificaciones de riesgo de LMA del estudio ALFA-0701 (población por IDTM)
| MYLOTARG + daunorubicina + citarabina | Daunorubicina + citarabina | |
| Citogenética (favorable/intermedia), N | 94 | 95 |
| Valor pc | < 0,0001 | |
| Citogenética (desfavorable), N | 27 | 30 |
| Valor pc | 0,7151 | |
| ELN (favorable/intermedia), N | 86 | 91 |
| Valor pc | 0,0003 | |
| ELN (desfavorable/adversa), N | 37 | 36 |
| Valor pc | 0,2091 |
El estudio ALFA-0701 no fue diseñado para evaluar prospectivamente el beneficio de MYLOTARG en subgrupos; los análisis se presentan solo con fines descriptivos.
Según la definición principal de SLE: fechas de los eventos (fallo de inducción, recaída o muerte) determinadas por la evaluación del investigador.
La población por IDTM incluyó a todos los pacientes aleatorizados, a menos que se retirara el consentimiento antes del inicio del tratamiento y se analizaron de acuerdo con el grupo de aleatorización inicial.
Abreviaturas: LMA = leucemia mieloide aguda; IC = intervalo de confianza; SLE = supervivencia libre de eventos; ELN = Red Europea de Leucemia; IDTM = intención de tratar modificada; n = número; N = número; NE = no estimable.
a. Mediana estimada usando el método Kaplan-Meier; IC basado en el método Brookmeyer-Crowley con transformación logarítmica en los dos ejes.
b. Según el modelo de riesgos proporcionales de Cox frente a daunorubicina + citarabina.
c. Valor p bilateral del test de log-rank.
Tabla 10. Supervivencia global conforme a las clasificaciones de riesgo de LMA del estudio ALFA-0701 (población por IDTM)
| MYLOTARG + daunorubicina + citarabina | Daunorubicina + citarabina | |
| Citogenética (favorable/intermedia), N | 94 | 95 |
| Valor pc | 0,1288 | |
| Citogenética (desfavorable), N | 27 | 30 |
| Valor pc | 0,1267 | |
| ELN (favorable/intermedia), N | 86 | 91 |
| Valor pc | 0,1216 | |
| ELN (desfavorable/adversa), N | 37 | 36 |
| Valor pc | 0,6487 |
El estudio ALFA-0701 no fue diseñado para evaluar prospectivamente el beneficio de MYLOTARG en subgrupos; los análisis se presentan sólo con fines descriptivos.
La población por IDTM incluyó a todos los pacientes aleatorizados, a menos que se retirara el consentimiento antes del inicio del tratamiento y se analizaron de acuerdo con el grupo de aleatorización inicial.
Abreviaturas: LMA = leucemia mieloide aguda; IC = intervalo de confianza; ELN = Red Europea de Leucemia;
IDTM = intención de tratar modificada; n = número; N = número; NE = no estimable; SG = supervivencia global.
d. Mediana estimada usando el método Kaplan-Meier; IC basado en el método Brookmeyer-Crowley con transformación logarítmica en los dos ejes.
e. Según el modelo de riesgos proporcionales de Cox frente a daunorubicina + citarabina.
f. Valor p bilateral del test de log-rank.
Población pediátrica
LMA no tratada previamente
En un estudio aleatorizado (COG AAML0531) que evaluó la quimioterapia habitual sola o combinada con MYLOTARG en 1 063 niños recién diagnosticados con LMA (93,7% de los pacientes < 18 años de edad) y adultos jóvenes (6,3% de los pacientes), con una mediana de edad de 8,9 años (intervalo: 0-29 años), los pacientes con LMA de novo fueron asignados de forma aleatoria o bien a solo quimioterapia habitual de 5 ciclos o bien a la misma quimioterapia con 2 dosis de MYLOTARG (3 mg/m2/dosis) administradas una vez en el ciclo de inducción 1 y una vez en el ciclo de intensificación 2. El estudio demostró que la adición de MYLOTARG a la quimioterapia intensiva mejoró la SLE (3 años: 50,6% frente al 44,0%; HR 0,838; IC del 95%: 0,706; 0,995; p = 0,0431) en la LMA de novo debido a un menor riesgo de recaída, con una tendencia hacia una mayor SG en el grupo de MYLOTARG que no fue estadísticamente significativa (3 años: 72,4% frente al 67,6%; HR 0,904; IC del 95%: 0,721; 1,133; p = 0,3799). Sin embargo, también se observó una mayor toxicidad (mortalidad tóxica posterior a la remisión) en pacientes con LMA de bajo riesgo, que se atribuyó a la neutropenia prolongada que se produjo después de recibir gemtuzumab ozogamicina durante el ciclo de intensificación 2 (ver secciones 4.2 y 4.8). En total, 29 (5,5%) pacientes en el grupo de MYLOTARG y 15 (2,8%) pacientes en el grupo del comparador murieron durante la remisión. Por lo tanto, no se estableció la dosis óptima de gemtuzumab ozogamicina para pacientes pediátricos (ver sección 4.2).
LMA recidivante o refractaria
Se realizó una revisión sistemática de la literatura médica de estudios para evaluar MYLOTARG en pacientes pediátricos con LMA recidivante o refractaria, que incluyó a 454 pacientes que habían recibido MYLOTARG en monoterapia (dosis únicas o fraccionadas) o un tratamiento combinado procedentes de 16 artículos publicados y del US Expanded Access Study (estudio de acceso ampliado de EEUU) (ver sección 4.8). La mediana del tamaño del estudio fue de 15 pacientes, con un intervalo de 5 a 105 pacientes. Las edades mínimas y máximas globales oscilaron entre 0 años y 22,3 años, y la mediana de edad global en el momento del tratamiento fue 8,7 años.
La mayoría de los estudios se realizaron en el ámbito del uso compasivo (70,6 %). MYLOTARG se administró en monoterapia en el 47,1 %, como parte de una combinación en el 23,5 % y en ambas modalidades en el 29,4 % de los estudios. La dosis total de MYLOTARG osciló entre 1,8 mg/m2 y 9 mg/m2. Cuando se administró MYLOTARG en combinación, se utilizó una pauta posológica a base de citarabina en 8 de los 9 estudios. En el 23,5 % de los estudios, la mayoría de los pacientes recibieron dosis fraccionadas (3 mg/m2 en los días 1, 4 y 7) de MYLOTARG, mientras que en el 35,3 % de los estudios se administraron dosis superiores a 3 mg/m2. MYLOTARG se administró como tratamiento de inducción en la mayoría de los estudios (82,4 %).
Con el tratamiento de MYLOTARG en monoterapia, la tasa de respuesta (RC/RCp/remisión completa con recuperación hematológica incompleta (RCi); media ponderada en los estudios) fue del 33,3 % con dosis fraccionadas (1 estudio) y del 24,3 % con dosis no fraccionadas (9 estudios). En la modalidad de combinación, la tasa de respuesta fue del 49,0 % con MYLOTARG no fraccionado (3 estudios) y del 38,8 % con MYLOTARG fraccionado (2 estudios).
La información de seguridad sobre mielosupresión, infecciones, EVO en general y EVO tras el TCMH y muerte, que son acontecimientos adversos conocidos de MYLOTARG (ver sección 4.8 y la tabla 6), se obtuvo de la literatura médica.
Las limitaciones de este análisis incluyen el tamaño pequeño de la muestra de algunos estudios, la heterogeneidad de los estudios y la falta de datos de control en este contexto.
Electrofisiología cardiaca
El efecto de MYLOTARG sobre el intervalo QT corregido se evaluó en el estudio en monoterapia B1761031, en 50 pacientes adultos con LMA CD33 positiva recidivante o refractaria. A concentraciones plasmáticas terapéuticas, la media del cambio máximo del intervalo QTcF con respecto al valor basal fue de 5,10 ms (IC del 90 %: 2,15; 8,06 ms). No hubo pacientes con un aumento máximo de QTcF con respecto al valor basal de > 60 ms y ningún paciente tuvo un QTcF > 480 ms. Se produjo un acontecimiento de fibrilación auricular (grado 3) y taquicardia supraventricular (grado 3) en el mismo paciente. No se notificaron acontecimientos adversos de arritmia cardiaca de grado 4 o grado 5.
Según el análisis de concentración-intervalo QTc, la mediana de cambio esperado en el QTcF con respecto al valor basal para el anticuerpo hP67.6 total a la Cmáx plasmática media observada, fue de 0,842 ms (IC del 95 %: −1,93; 3,51 ms). Para la calicheamicina no conjugada, la mediana del cambio esperado en el QTcF con respecto al valor basal fue de 0,602 ms (IC del 95 %: −2,17; 2,72 ms) a la Cmáx plasmática aproximada observada tras la administración con la pauta posológica recomendada de MYLOTARG.
5.2 - Propiedades farmacocinéticas de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Gemtuzumab ozogamicina es un conjugado de anticuerpo y fármaco (CAF) compuesto por un anticuerpo monoclonal dirigido contra CD33 (hP67.6) que está unido covalentemente al citotóxico N-acetil-gamma-calicheamicina. La farmacocinética (FC) de gemtuzumab ozogamicina se describe midiendo las características FC del anticuerpo (hP67.6), así como de los derivados de calicheamicina conjugados y no conjugados.
Se recopilaron datos clínicos de FC tras una pauta posológica de monoterapia (3 mg/m2 hasta un máximo de un vial de 5 mg los días 1, 4 y 7) de MYLOTARG. Las exposiciones medidas según la media geométrica del AUC336 y la Cmáx después de dosis múltiples para calicheamicina conjugada y anticuerpo hP67.6 total fueron de 461 500 pg·hr/ml y 11 740 pg/ml, y 26 820 ng·hr/ml y 585,6 ng/ml, respectivamente. Los datos de FC para la calicheamicina no conjugada no se han presentado debido a problemas de inestabilidad en el plasma.
Distribución
In vitro, la unión de N-acetil-gamma-calicheamicina dimetilhidrazida a las proteínas plasmáticas humanas es de aproximadamente el 97%. In vitro, N-acetil-gamma-calicheamicina dimetilhidrazida es un sustrato de la glicoproteína P (P-gp). En los pacientes, el volumen total de distribución del anticuerpo hP67.6 (suma de V1 [13,0 litros] y V2 [6,91 litros]) fue de aproximadamente 20 litros.
Biotransformación
Se prevé que la vía metabólica principal de gemtuzumab ozogamicina sea la liberación hidrolítica de N-acetil-gamma-calicheamicina dimetilhidrazida. Los estudios in vitro demostraron que N-acetil-gamma-calicheamicina dimetilhidrazida se metaboliza considerablemente, principalmente a través de la reducción no enzimática del grupo disulfuro. Se prevé que la actividad (citotoxicidad) de los metabolitos resultantes se atenúe significativamente.
Interacciones con otros medicamentos
Efecto de otros medicamentos sobre gemtuzumab ozogamicina
In vitro, N-acetil-gamma-calicheamicina dimetilhidrazida se metaboliza de forma principal a través de una reducción no enzimática. Por lo tanto, es poco probable que la administración concomitante de gemtuzumab ozogamicina con inhibidores o inductores del citocromo P450 (CYP) o de enzimas metabolizadoras de la uridina difosfato glucuronosiltransferasa (UGT) altere la exposición a N-acetil-gamma-calicheamicina dimetilhidrazida.
Según los análisis de farmacocinética (FC) poblacional, no se prevé que la combinación de gemtuzumab ozogamicina con hidroxiurea, DNR y AraC cause cambios clínicamente significativos en la FC de hP67.6 o de la calicheamicina no conjugada.
Efecto de gemtuzumab ozogamicina sobre otros medicamentos
Efecto sobre los sustratos de CYP
In vitro, N-acetil-gamma-calicheamicina dimetilhidrazida y gemtuzumab ozogamicina presentaron un bajo potencial para inhibir las actividades de CYP1A2, CYP2A6 (analizadas únicamente con gemtuzumab ozogamicina), CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4/5 a concentraciones clínicamente relevantes. In vitro, N-acetil-gamma-calicheamicina dimetilhidrazida y gemtuzumab ozogamicina presentaron un bajo potencial para inducir las actividades de CYP1A2, CYP2B6 y CYP3A4 a concentraciones clínicamente relevantes.
Efecto sobre los sustratos de UGT
In vitro, N-acetil-gamma-calicheamicina dimetilhidrazida presentó un bajo potencial para inhibir las actividades de UGT1A1, UGT1A4, UGT1A6, UGT1A9 y UGT2B7 a concentraciones clínicamente relevantes.
Efecto sobre los sustratos del transportador de fármacos
In vitro, N-acetil-gamma-calicheamicina dimetilhidrazida presentó un bajo potencial para inhibir las actividades de la P-gp, la proteína de resistencia al cáncer de mama (BCRP, por sus siglas en inglés), la bomba de exportación de sales biliares (BSEP, por sus siglas en inglés), la proteína relacionada con resistencia a múltiples fármacos (MRP, por sus siglas en inglés) 2, la proteína de extrusión de multifármacos y toxinas (MATE, por sus siglas en inglés)1 y MATE2K, el transportador de aniones orgánicos (OAT, por sus siglas en inglés)1 y OAT3, el transportador de cationes orgánicos (OCT, por sus siglas en inglés)1 y OCT2 y el polipéptido transportador de aniones orgánicos (OATP, por sus siglas en inglés)1B1 y OATP1B3 a concentraciones clínicamente relevantes.
Efecto sobre medicamentos quimioterapéuticos coadministrados
Según los análisis de farmacocinética (FC) poblacional, no se prevé que la combinación de gemtuzumab ozogamicina con DNR y AraC produzca cambios clínicamente significativos en la FC de estos medicamentos.
Eliminación
La FC de gemtuzumab ozogamicina está bien caracterizada mediante un modelo bicompartimental con componentes de eliminación lineales y dependientes del tiempo. En 50 pacientes con LMA recidivante o refractaria tras una pauta posológica en monoterapia (3 mg/m2 hasta un máximo de un vial de 5 mg los días 1, 4 y 7) de MYLOTARG, la eliminación del anticuerpo hP67.6 total fue de 0,288 l/h, y la semivida (t½) de eliminación terminal se estimó en 96,6 h.
Farmacocinética en grupos específicos de personas o pacientes
Edad, raza y sexo
Según un análisis de FC poblacional, la edad, la raza y el sexo no afectaron de forma significativa a la disposición de gemtuzumab ozogamicina.
Insuficiencia hepática
No se han realizado estudios formales de FC con gemtuzumab ozogamicina en pacientes con insuficiencia hepática.
Según un análisis de FC poblacional, no se espera que el aclaramiento de gemtuzumab ozogamicina (anticuerpo hP67.6 y calicheamicina no conjugada) se vea afectado por un estado de insuficiencia hepática leve, según lo definido por el Grupo de trabajo de Disfunción Orgánica del Instituto Nacional del Cáncer de los EE.UU. (NCI ODWG, por sus siglas en inglés). El análisis incluyó a 405 pacientes en las siguientes categorías de estado de insuficiencia del NCI ODWG: leve (B1, n = 58 y B2, n = 19), moderada (C, n = 6) y función hepática normal (n = 322) (ver sección 4.2).
Insuficiencia renal
No se han realizado estudios formales de FC con gemtuzumab ozogamicina en pacientes con insuficiencia renal.
Según un análisis FC poblacional en 406 pacientes, la eliminación de gemtuzumab ozogamicina en pacientes con insuficiencia renal leve (aclaramiento de creatinina [CLcr] 60-89 ml/min; n = 149) o insuficiencia renal moderada (CLcr 30-59 ml/min; n = 47) fue similar a la de los pacientes con una función renal normal (CLcr≥90 ml/min; n = 209). La FC de gemtuzumab ozogamicina no se ha estudiado en pacientes con insuficiencia renal grave.
Población pediátrica
Los resultados del modelo poblacional mostraron que el comportamiento FC de gemtuzumab ozogamicina (anticuerpo hP67.6 y calicheamicina no conjugada) es similar entre los pacientes adultos y pediátricos con LMA después de la pauta posológica de 9 mg/m2.
5.3 - Datos preclínicos sobre seguridad de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Toxicidad a dosis repetidas
Las principales toxicidades se produjeron en hígado, médula ósea y órganos linfoides, en los parámetros hematológicos (disminución de la masa de glóbulos rojos y de los recuentos de leucocitos, principalmente linfocitos), riñón, ojos y órganos reproductores masculinos y femeninos. Los efectos en hígado, riñón y órganos reproductores masculinos en ratas y en tejidos linfoides en monos (aproximadamente 18 veces para ratas y 36 veces para monos la exposición clínica en humanos después de la tercera dosis para humanos de 3 mg/m2 según el AUC168) fueron irreversibles. Los efectos sobre los órganos reproductores femeninos y los ojos en monos fueron adversos en el estudio de 12 semanas (aproximadamente 193 y 322 veces, respectivamente, la exposición clínica en humanos después de la tercera dosis para humanos de 3 mg/m2 según el AUC168). Se desconoce la relevancia de los hallazgos irreversibles en animales para los seres humanos. No se observaron efectos en el sistema nervioso en animales después de la administración de MYLOTARG. Se identificaron alteraciones del sistema nervioso en ratas con otros conjugados anticuerpo-calicheamicina.
Genotoxicidad
Gemtuzumab ozogamicina resultó ser clastogénico. Esto es coherente con la conocida inducción de roturas en el ADN producidas por la calicheamicina y otros antibióticos antitumorales enodiinos. N-acetil-gamma-calicheamicina DMH (la citotoxina liberada) resultó ser mutagénica y clastogénica.
Carcinogenicidad
No se han realizado estudios formales de potencial carcinogénico con gemtuzumab ozogamicina. En los estudios de toxicidad, las ratas presentaron lesiones preneoplásicas (hiperplasia de células ovales de mínima a leve) en el hígado a aproximadamente 54 veces la exposición clínica en humanos después de la tercera dosis para humanos de 3 mg/m2 según el AUC168. No se observaron lesiones preneoplásicas o neoplásicas en monos hasta aproximadamente 115 veces la exposición clínica en humanos después de la tercera dosis para humanos de 3 mg/m2 según el AUC168. Se desconoce la relevancia de estos hallazgos en animales para los seres humanos.
Toxicidad para la reproducción
En un estudio de fertilidad en ratas hembra se observaron cantidades ligeramente menores de cuerpos lúteos y aumento de la embrioletalidad en presencia de toxicidad materna (aproximadamente 9,7 veces la exposición clínica en humanos después de la tercera dosis para humanos de 3 mg/m2 según el AUC168). Se observaron efectos en los órganos reproductores de monos hembra en el estudio de 12 semanas (atrofia de ovarios, oviducto, útero y cuello uterino, aproximadamente 193 veces la exposición clínica en humanos después de la tercera dosis de 3 mg/m2).
En un estudio de fertilidad masculina, los efectos sobre la reproducción masculina incluyeron espermatogonias y espermatocitos reducidos, disminución de espermátides testiculares y espermatozoides epididimarios, vacuolación del núcleo en las espermátides y/o aparición de células gigantes. Otros hallazgos incluyeron efectos sobre los testículos, los epidídimos y las glándulas mamarias, así como la fertilidad. Cuando las ratas macho se aparearon de nuevo después de un periodo sin tratamiento de 9 semanas, los efectos sobre el esperma y la fertilidad fueron más graves, pero hubo una recuperación parcial de la reducción de las espermatogonias y los espermatocitos en los testículos. Los efectos sobre los órganos reproductores de ratas macho fueron parcialmente reversibles o irreversibles (ver sección 4.6). Se observaron efectos en los órganos reproductores de monos macho (testículos, epidídimos, vesículas seminales) a aproximadamente 66 veces la exposición clínica en humanos después de la tercera dosis de 3 mg/m2.
En un estudio de toxicidad embriofetal, se observó un menor peso corporal fetal, una mayor incidencia de costillas onduladas fetales y una menor incidencia de osificación esquelética fetal. El aumento de la embrioletalidad y las anomalías morfológicas fetales incluyeron malformaciones digitales, ausencia del arco aórtico, anomalías en los huesos largos en las extremidades anteriores, omóplato deformado, ausencia de centro vertebral y esternones fusionados. El aumento de la embrioletalidad también se observó en presencia de toxicidad materna. La dosis más baja con efectos embriofetales se correlacionó con 9,7 veces la exposición clínica en humanos después de la tercera dosis para humanos de 3 mg/m2 según el AUC168 (ver sección 4.6).
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Dextrano 40
Sacarosa
Cloruro de sodio
Dihidrógeno fosfato de sodio monohidrato
Fosfato disódico de hidrógeno anhidro
6.2 - Incompatibilidades de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 - Período de validez de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Vial sin abrir
5 años
Solución reconstituida y diluida
Proteger las soluciones reconstituidas y diluidas de MYLOTARG de la luz. Las soluciones se deben utilizar de forma inmediata. No congelar la solución reconstituida o diluida.
Si el medicamento no se puede utilizar de forma inmediata:
- Tras la reconstitución, el vial original se puede conservar hasta un máximo de 16 horas en nevera (entre 2°C y 8°C) o hasta un máximo de 3 horas a temperatura ambiente (por debajo de 30°C).
- La solución diluida se puede conservar hasta un máximo de 18 horas en nevera (entre 2°C y 8°C) y hasta un máximo de 6 horas a temperatura ambiente (por debajo de 30°C). El tiempo permitido a temperatura ambiente (por debajo de 30°C) incluye el tiempo requerido para la preparación de la solución diluida, equilibrado, si es necesario, y administración al paciente. El tiempo máximo desde la preparación de la solución diluida hasta la administración no debe exceder las 24 horas.
6.4 - Precauciones especiales de conservación de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación tras la reconstitución y dilución del medicamento, ver sección 6.3.
6.5 - Naturaleza y contenido del recipiente de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Vial de vidrio ámbar tipo I con tapón de goma de butilo y cápsula de cierre de tipo flip-off conteniendo 5 mg de gemtuzumab ozogamicina.
Cada envase contiene 1 vial.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de MYLOTARG 5 mg Polvo concentrado para sol. para perfus.
Utilizar una técnica aséptica apropiada para los procedimientos de reconstitución y dilución. MYLOTARG es sensible a la luz y se debe proteger de la luz ultravioleta durante la reconstitución, dilución y administración.
Reconstitución
- Calcular la dosis (mg) de MYLOTARG requerida.
- Antes de la reconstitución, dejar que el vial alcance la temperatura ambiente (por debajo de 30°C) durante, aproximadamente, 5 minutos. Reconstituir cada vial de 5 mg con 5 ml de agua para preparaciones inyectables para obtener una solución de un solo uso de 1 mg/ml de gemtuzumab ozogamicina.
- Mover suavemente el vial para favorecer la disolución. No agitar.
- Inspeccionar la solución reconstituida en busca de partículas o decoloración. La solución reconstituida puede contener partículas pequeñas de color blanco a blanquecino, de opacas a translúcidas y sin forma definida o con una forma similar a las fibras.
- MYLOTARG no contiene conservantes bacteriostáticos.
- Si la solución reconstituida no se puede utilizar de forma inmediata, se puede conservar en el vial original hasta un máximo de 16 horas en nevera (entre 2°C y 8°C) o hasta un máximo de 3 horas a temperatura ambiente (por debajo de 30°C). Proteger de la luz y no congelar.
Dilución
- Calcular el volumen requerido de la solución reconstituida necesaria para obtener la dosis adecuada en función de la superficie corporal del paciente. Extraer esta cantidad del vial con una jeringa. Los viales de MYLOTARG contienen 5 mg de medicamento sin sobrellenado. Cuando se reconstituye a una concentración de 1 mg/ml según las indicaciones, el contenido extraíble del vial es de 4,5 mg (4,5 ml). Proteger de la luz. Desechar cualquier solución reconstituida no utilizada que quede en el vial.
- Las dosis se deben mezclar a una concentración de entre 0,075 mg/ml y 0,234 mg/ml según las siguientes instrucciones:
- Las dosis inferiores a 3,9 mg se deben preparar para la administración con una jeringa. Agregar la solución reconstituida de MYLOTARG a una jeringa con solución inyectable de cloruro sódico 9 mg/ml (0,9%) hasta una concentración final de entre 0,075 mg/ml y 0,234 mg/ml. Proteger de la luz.
- Las dosis superiores o iguales a 3,9 mg se deben diluir en una jeringa o una bolsa intravenosa con un volumen apropiado de solución inyectable de cloruro sódico 9 mg/ml (0,9%) para garantizar una concentración final de entre 0,075 mg/ml y 0,234 mg/ml. Proteger de la luz.
- Invertir suavemente el recipiente de perfusión para mezclar la solución diluida. No agitar.
- Tras la dilución con la solución inyectable de cloruro sódico 9 mg/ml (0,9%), la solución de MYLOTARG se debe perfundir de forma inmediata. Si no se utiliza de inmediato, la solución diluida se puede conservar hasta 18 horas en nevera (entre 2°C y 8°C) y hasta un máximo de 6 horas a temperatura ambiente (por debajo de 30°C). El tiempo permitido a temperatura ambiente (por debajo de 30°C) incluye el tiempo requerido para la preparación de la solución diluida, equilibrado, si es necesario, y administración al paciente. El tiempo máximo desde la preparación de la solución diluida hasta la administración no debe exceder las 24 horas. Proteger de la luz y no congelar.
- Se recomienda que el recipiente de perfusión esté hecho de cloruro de polivinilo (PVC, por sus siglas en inglés) con DEHP, etilvinilacetato (EVA) o poliolefina (polipropileno y/o polietileno).
Administración
- Se requiere la filtración de la solución diluida. Para la perfusión de MYLOTARG se utilizará un filtro de poliétersulfona (PES) de 0,2 micras de baja unión a proteínas en línea.
- Las dosis administradas con una jeringa deben utilizar líneas de perfusión de pequeño calibre (microbore) con un filtro de poliétersulfona (PES) de 0,2 micras de baja unión a proteínas en línea.
- Durante la perfusión, la bolsa intravenosa o las jeringas deben protegerse de la luz (incluida la luz ultravioleta) con una cubierta que bloquee la entrada de la luz. No es necesario proteger de la luz la línea de perfusión.
- Perfundir la solución diluida durante 2 horas. La perfusión se debe completar antes de que finalice el periodo de conservación permitido de 6 horas de la solución diluida a temperatura ambiente (por debajo de 30°C).
- Se recomienda que las líneas de perfusión estén hechas de PVC (con o sin DEHP), poliuretano o polietileno.
No mezclar ni administrar MYLOTARG en perfusión con otros medicamentos.
Consultar también la sección 6.3 para obtener información sobre la dilución, la conservación y la perfusión.
Eliminación
Se deben utilizar los procedimientos de eliminación de desechos tóxicos establecidos para los medicamentos contra el cáncer.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Bruxelles
Bélgica
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/18/1277/001
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19/abril/2018
Fecha de la última renovación:
10. - FECHA DE LA REVISIÓN DEL TEXTO
11/2022
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
PRESENTACIONES Y PRECIO
MYLOTARG 5 mg polvo para concentrado para solución para perfusión, envase con 1 vial. PVL: 7.200,00€; PVP: 7.255,91€; PVP IVA: 7.546,15€.
CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN
Con receta médica. Uso Hospitalario.
CONDICIONES DE LA PRESTACIÓN FARMACÉUTICA
Financiado por el Sistema Nacional de Salud, sin aportación. Información disponible en la web del Ministerio de Sanidad https://www.mscbs.gob.es/home.htm.
Para información adicional, por favor, contacte con el Centro de Información Médico-Farmacéutica llamando al + 34 914909900 o consulte nuestra página web www.pfizer.es.

