

Mecanismo de acciónLevetiracetam
Reduce la liberación de Ca<exp>2+<\exp> intraneuronal y se une a la proteína 2A de las vesículas sinápticas, involucrada en la exocitosis de neurotransmisores.
Indicaciones terapéuticasLevetiracetam
- Monoterapia: tto. de crisis de inicio parcial con/sin generalización secundaria en ads. y adolescentes de 16 años de edad o mayores con un nuevo diagnóstico de epilepsia..
- Terapia concomitante: tto. de crisis de inicio parcial con/sin generalización secundaria en ads. y niños > 1 mes con epilepsia; en crisis mioclónicas en ads. y niños > 12 años con epilepsia mioclónica juvenil y crisis tónico-clónicas generalizadas primarias en ads. y adolescentes > 12 años con epilepsia generalizada idiopática.
PosologíaLevetiracetam
oral. La solución oral es la formulación más adecuada para su administración en lactantes y niños < 6 años. Se encuentra disponible en en tres formatos: frasco de 150 ml con jeringa de 1 ml, frasco de 150 ml con jeringa de 3 ml y frasco de 300 ml con jeringa de 10 ml, se han notificado casos de sobredosis accidental en niños en los que llegaron a administrase hasta 10 veces la dosis pautada, debido a confusión de la dosis administrada o a que la jeringa que se empleó para dosificar no era la correcta. Para tratar de evitar que se produzcan más errores de dosificación, se ha instado a los titulares de la autorización de comercialización que comercializan más de una presentación de levetiracetam solución oral a usar códigos de color y pictogramas para: diferenciar una presentación de otra, indicar claramente el rango de edad para el cual está prevista cada presentación e indicar claramente en la caja/etiquetado qué dispositivo de dosificación debe utilizarse con cada presentación concreta. Se realizará un continuo seguimiento de tales medidas.
- Monoterapia: ads. y > 16 años: inicial, 250 mg/12 h, tras 2 sem aumentar a 500 mg/12 h; aumentar según respuesta, 250 mg/12 h cada 2 sem hasta máx. 1.500 mg/12 h.
- Terapia concomitante: ads. y niños > 12 años (p.c. >= 50 kg): inicial, 500 mg/12 h; aumentar según respuesta, 500 mg/12 h cada 2-4 sem hasta máx. 1.500 mg/12 h. Lactantes de 6 -23 meses, niños (2 - 11 años) y adolescentes (12- 17 años) con un p.c.< 50 kg: inicial, 10 mg/kg/12 h; aumentar según respuesta 10 mg/kg/12 h cada 2 sem hasta máx. 30 mg/kg/12 h. Lactantes desde 1 mes a < de 6 meses: 7 mg/kg/12 h; aumentar según respuesta 7 mg/kg/12 h cada 2 sem hasta máx. 21 mg/kg/12 h.
I.R.:
- Clcr 50-79 ml/min: ads. y adolescentes >50 kg: 500-1.000 mg/12 h; lactantes 1 a < 6 meses:7-14 mg/kg/12 h; lactantes de 6-23 meses niños y a adolescentes de < 50 kg: 10-20 mg/kg/12 h.
- Clcr 30-49 ml/min: ads. y adolescentes >50 kg: 250-750 mg/12 h; lactantes 1 a < 6 meses: 3,5-10,5 mg/kg/12 h; lactantes de 6-23 meses niños y a adolescentes de < 50 kg: 5-15 mg/kg/12 h.
- Clcr < 30 ml/min: ads. y adolescentes >50 kg: 250-500 mg/12 h; lactantes 1 a < 6 meses:3,5-7 mg/kg/12 h; lactantes de 6-23 meses niños y a adolescentes de < 50 kg:5-10 mg/kg/12 h. Dializados con enf. renal: ads. y adolescentes >50 kg: 500-1.000 mg/24 h, con dosis de carga 750 mg el 1<exp>er<\exp> día y 250-500 mg después de la diálisis; lactantes 1 a < 6 meses: 7-14 mg/kg/día con dosis de carga de 10,5 mg/kg el 1<exp>er<\exp> día y 3,5-7 mg/kg después de la diálisis; lactantes de 6-23 meses niños y a adolescentes de < 50 kg:10-20 mg/kg con dosis de carga de 15 mg/kg el 1<exp>er<\exp> día y 5-10 mg/kg después de la diálisis.
IV.
Alternativa cuando administración oral no es viable temporalmente. Perfus. IV de 15 min.
- Monoterapia ads. y adolescentes > 16 años: inicial:250 mg/12 h, aumentar hasta 500 mg/12 h, tras 2 sem de tto. Aumentar en función de la respuesta clínica con 250 mg/12 h cada 2 semanas. La dosis máxima es de 1500 mg/12 h. No se ha establecido la seguridad y eficacia en niños y adolescentes <16 años.
- Terapia concomitante: ads. y niños > 12 años (p.c. >= 50 kg): inicial: 500 mg/12 h. Esta dosis se puede instaurar desde el primer día de tto. Dependiendo de la respuesta clínica y de la tolerabilidad, incrementar hasta 1500 mg /12 h. La modificación de la dosis se puede realizar con aumentos o reducciones de 500 mg /12 h cada 2 a 4 sem.
Niños 4-11 años y adolescentes (12-17 años) (p.c.< 50 kg): inicial : 10 mg/kg /12 h. En función de la respuesta clínica y tolerabilidad, aumentar hasta3 0 mg/kg /12 h. Los cambios de dosis no deben exceder de aumentos o reducciones de 10 mg/kg /12 h cada dos semanas. No se ha establecido la seguridad y eficacia en niños <4 años.
No hay experiencia por periodo > 4 días.
Modo de administraciónLevetiracetam
Vía oral. Los comprimidos recubiertos con película se administran con una cantidad suficiente de líquido y pueden administrarse con o sin alimentos. La posología diaria se divide en dosis iguales repartidas en dos tomas al día. La solución oral puede diluirse en un vaso de agua o en un biberón y puede administrarse con o sin alimentos.
Vía IV. Concentrado para solución para perfusión: diluir la dosis recomendada en 100 ml de un diluyente compatible como mínimo y administrarse por vía IV como una perfusión IV de 15 minutos. Solución para perfusión: la dosis recomendada debe administrarse como una perfusión IV de 15 minutos.
ContraindicacionesLevetiracetam
Hipersensibilidad a levetiracetam, a otros derivados de pirrolidona.
Advertencias y precaucionesLevetiracetam
I.R., I.H. grave. Se han notificado casos de suicidio, intento y pensamientos y comportamientos suicidas. Suspender gradualmente tto. La formulación en comprimidos no está adaptada para su administración en lactantes y niños < 6 años, la solución oral es la formulación más adecuada para su administración en esta población. Riesgo de lesión renal aguda. Riesgo de neutropenia, agranulocitosis, leucopenia, trombocitopenia y pancitopenia, si aparece debilidad importante, pirexia, infecciones recurrentes o trastornos de la coagulación, realizar recuento de células sanguíneas completo.
Insuficiencia hepáticaLevetiracetam
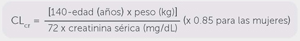
Precaución. En I.H. grave, el aclaramiento de creatinina puede subestimar el grado de I.R. Por lo tanto, se recomienda una reducción del 50 % de la dosis de mantenimiento diario cuando el Clcr < 60 ml/min.
Insuficiencia renalLevetiracetam
Precaución. Ajustar posología: oral:
- Clcr 50-79 ml/min: das. y adolescentes >50 kg: 500-1.000 mg/12 h; lactantes 1 a < 6 meses:7-14 mg/kg/12 h; lactantes de 6-23 meses niños y a adolescentes de < 50 kg: 10-20 mg/kg/12 h.
- Clcr 30-49 ml/min: das. y adolescentes >50 kg: 250-750 mg/12 h; lactantes 1 a < 6 meses: 3,5-10,5 mg/kg/12 h; lactantes de 6-23 meses niños y a adolescentes de < 50 kg: 5-15 mg/kg/12 h.
- Clcr < 30 ml/min: das. y adolescentes >50 kg: 250-500 mg/12 h; lactantes 1 a < 6 meses:3,5-7 mg/kg/12 h; lactantes de 6-23 meses niños y a adolescentes de < 50 kg:5-10 mg/kg/12 h. Dializados con enf. renal: das. y adolescentes >50 kg: 500-1.000 mg/24 h, con dosis de carga 750 mg el 1<exp>er<\exp> día y 250-500 mg después de la diálisis; lactantes 1 a < 6 meses: 7-14 mg/kg/día con dosis de carga de 10,5 mg/kg el 1<exp>er<\exp> día y 3,5-7 mg/kg después de la diálisis; lactantes de 6-23 meses niños y a adolescentes de < 50 kg:10-20 mg/kg con dosis de carga de 15 mg/kg el 1<exp>er<\exp> día y 5-10 mg/kg después de la diálisis.
InteraccionesLevetiracetam
Aumenta la concentración plasmática de: metotrexato, vigilar los niveles plasmáticos de metotrexato y levetiracetam.
Eficacia disminuida (vía oral) con: macrogol, no se debe tomar macrogol por vía oral al menos durante una hora antes o una hora después de tomar levetiracetam.
EmbarazoLevetiracetam
El tratamiento con múltiples medicamentos antiepilépticos está asociado con un mayor riesgo de malformaciones congénitas que la monoterapia y por tanto se debe considerar la monoterapia. Los estudios en animales han mostrado toxicidad reproductiva. No se recomienda durante el embarazo ni en mujeres en edad fértil que no utilicen métodos anticonceptivos efectivos, a menos que sea clínicamente necesario. Al igual que con otros medicamentos antiepilépticos, los cambios fisiológicos durante el embarazo pueden afectar a las concentraciones de levetiracetam. Se ha observado la disminución de las concentraciones plasmáticas de levetiracetam durante el embarazo. Esta disminución es más pronunciada durante el tercer trimestre (hasta el 60% de la concentración inicial antes del embarazo). Debe asegurarse un control clínico adecuado de la mujer embarazada tratada con levetiracetam. La retirada de los tratamientos antiepilépticos puede dar lugar a una exacerbación de la enfermedad, que podría perjudicar a la madre y al feto.
LactanciaLevetiracetam
Levetiracetam se excreta en la leche materna humana, por lo que no se recomienda la lactancia natural. Sin embargo, si durante el periodo de lactancia es necesario el tratamiento con levetiracetam, debe considerarse la relación beneficio/riesgo del tratamiento teniéndose en cuenta la importancia de la lactancia natural.
Efectos sobre la capacidad de conducirLevetiracetam
Levetiracetam actúa sobre el sistema nervioso central y puede producir: somnolencia, mareos, alteraciones visuales y disminución de la capacidad de reacción. Estos efectos así como la propia enfermedad hacen que sea recomendable tener precaución a la hora de conducir vehículos o manejar maquinaria peligrosa, especialmente mientras no se haya establecido la sensibilidad particular de cada paciente al medicamento.
Reacciones adversasLevetiracetam
Nasofaringitis; anorexia; depresión, hostilidad/agresividad, ansiedad, nerviosismo/irritabilidad; somnolencia, cefalea, convulsión, trastorno del equilibrio, mareo, letargo, temblor; vértigo; tos; dolor abdominal, diarrea, dispepsia, vómitos, náuseas; rash; astenia/fatiga.
© Vidal Vademecum Fuente: El contenido de esta monografía de principio activo según la clasificación ATC, ha sido redactado teniendo en cuenta la información clínica de todos los medicamentos autorizados y comercializados en España clasificados en dicho código ATC. Para conocer con detalle la información autorizada por la AEMPS para cada medicamento, deberá consultar la correspondiente Ficha Técnica autorizada por la AEMPS.
Monografías Principio Activo: 06/11/2018