OPTIMARK 500 MICROMOL/ml SOLUCION INYECTABLE EN UN VIAL
| ATC: Gadoversetamida |
| PA: Gadoversetamida |
Envases
- Env. con 10 viales de 10 ml
- H: Medicamento de uso hospitalario
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica- Facturable SNS: NO
- Comercializado: No
- Situación: Anulado
- Código Nacional: 602703
- EAN13: 8470006027035
- Env. con 10 viales de 15 ml
- H: Medicamento de uso hospitalario
- Dispensación sujeta a prescripción médica
- Facturable SNS: NO
- Comercializado: No
- Situación: Anulado
- Código Nacional: 602704
- EAN13: 8470006027042
- Env. con 10 viales de 20 ml
- H: Medicamento de uso hospitalario
- Dispensación sujeta a prescripción médica
- Facturable SNS: NO
- Comercializado: No
- Situación: Anulado
- Código Nacional: 602705
- EAN13: 8470006027059
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
OPTIMARK 500 mcmol/ml Sol. iny. en vial
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Jeringa precargada
1 ml contiene 330,9 mg de gadoversetamida, lo que equivale a 500 micromol.
Cada jeringa de 10 ml contiene 3309 mg de gadoversetamida, equivalente a 5 milimol. Cada jeringa de 15 ml contiene 4963,5 mg de gadoversetamida, equivalente a 7,5 milimol. Cada jeringa de 20 ml contiene 6618 mg de gadoversetamida, equivalente a 10 milimol. Cada jeringa de 30 ml contiene 9927 mg de gadoversetamida, equivalente a 15 milimol.
Excipiente(s) con efecto conocido:
20 ml de solución contienen 28,75 mg de sodio.
30 ml de solución contienen 43,13 mg de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1. Vial
1 ml contiene 330,9 mg de gadoversetamida, lo que equivale a 500 micromol.
Cada vial de 10 ml contiene 3309 mg de gadoversetamida, equivalente a 5 milimol. Cada vial de 15 ml contiene 4963,5 mg de gadoversetamida, equivalente a 7,5 milimol. Cada vial de 20 ml contiene 6618 mg de gadoversetamida, equivalente a 10 milimol.
Excipiente(s) con efecto conocido:
20 ml de solución contienen 28,75 mg de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. - FORMA FARMACÉUTICA
Jeringa precargada
Solución inyectable en jeringa precargada.
Vial
Solución inyectable en vial.
Solución transparente, de incolora a amarillo pálido pH: 6,0 – 7,5
Osmolaridad (37°C): 1000 – 1200 mOsm/kg
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Este medicamento es únicamente para uso diagnóstico.
Optimark está indicado para utilización en imagen por resonancia magnética (RM) del sistema nervioso central (SNC) e hígado. Intensifica el contraste y facilita la visualización, ayudando a la caracterización de lesiones focales y estructuras anormales en el SNC e hígado en pacientes adultos y en niños de dos o más años de edad con patología conocida o sospechada.
4.2 - Posología y administración de OPTIMARK 500 mcmol/ml Sol. iny. en vial
4.3 - Contraindicaciones de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Hipersensibilidad a la gadoversetamida o a otros productos que contienen gadolinio o a alguno de los excipientes incluidos en la sección 6.1.
Optimark está contraindicado
• en pacientes con insuficiencia renal grave (tasa de filtración glomerular TFG <30 ml/min/1,73 m²)
y/o lesión renal aguda,
• en pacientes que se hayan sometido a un trasplante hepático o
• que se encuentren en el período perioperatorio de un trasplante hepático y
• en recién nacidos de hasta 4 semanas de edad (ver sección 4.4).
4.4 - Advertencias y Precauciones de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Al igual que con otros medios de contraste paramagnéticos, el realce del contraste de la imagen de RM con gadoversetamida puede empeorar la visualización de lesiones existentes. Algunas de estas lesiones pueden detectarse mediante RM sin contraste. Por lo tanto, se debe actuar con precaución cuando la interpretación de la imagen realzada de contraste se realice en ausencia de una imagen de RM sin contraste
Antes de la prueba, los pacientes deben estar suficientemente hidratados.
Hipersensibilidad
También pueden ocurrir reacciones de tipo alérgico y otras relaciones idiosincrásicas con gadoversetamida, lo que podría manifestarse en forma de reacciones cardiovasculares, respiratorias y cutáneas (ver sección 4.8). La mayoría de estas reacciones ocurren en la primera media hora tras la administración del medio de contraste. Como ocurre con todos los demás medios de contraste de la misma clase, pueden producirse raramente reacciones tardías (después de horas o días) en casos raros; sin embargo, no se ha notificado ningún caso en los ensayos clínicos realizados.
Si se producen reacciones de hipersensibilidad, la administración del medio de contraste debe interrumpirse inmediatamente e iniciar un tratamiento intravenoso, si es necesario.
La prueba debe realizarse bajo la supervisión de un médico y se recomienda la inserción de un catéter flexible para acceso intravenoso. Para permitir una acción inmediata en caso de emergencia, debe garantizarse la disponibilidad inmediata de los medicamentos necesarios (por ejemplo, epinefrina/adrenalina, teofilina, antihistamínicos, corticosteroides y atropinas), tubo endotraqueal y
ventilador.
El riesgo de reacciones de hipersensibilidad aumenta en los siguientes casos:
- pacientes con predisposición alérgica
- pacientes con asma bronquial; en estos pacientes es el riesgo de broncoespasmo lo que aumenta particularmente
- pacientes con un historial de reacciones a medios de contraste, incluyendo los medios yodados Antes de la inyección del medio de contraste, debe preguntarse a los pacientes si tienen alguna alergia (por ejemplo alergias al pescado o a medicamentos, fiebre del heno, urticaria) si son hipersensibles a
los medios de contraste y si padecen asma bronquial. Puede considerarse una premedicación con antihistamínicos y/o glucocorticoides.
Pacientes en tratamiento con betabloqueantes
Se debe tener en cuenta que los pacientes que utilizan beta-bloqueantes no responden necesariamente a los beta-agonistas utilizados habitualmente para el tratamiento de las reacciones de hipersensibilidad.
Pacientes con trastornos cardiovasculares
En este grupo de pacientes las reacciones de hipersensibilidad pueden ser graves. Especialmente en pacientes con enfermedades cardíacas graves (por ejemplo, insuficiencia cardiaca grave, enfermedad arterial coronaria), las reacciones cardiovasculares pueden empeorar. Sin embargo, no hubo evidencias en los ensayos clínicos con Optimark.
Trastornos del sistema nervioso central
En pacientes con epilepsia o lesiones cerebrales puede aumentar la probabilidad de sufrir convulsiones durante la prueba. Son necesarias precauciones al realizar la exploración de estos pacientes (por ejemplo vigilancia del paciente), y debe estar disponible el equipo y los medicamentos necesarios para el tratamiento rápido de las posibles convulsiones.
Pacientes con insuficiencia renal
Antes de administrar Optimark, debe evaluarse a todos los pacientes para descartar disfunción renal mediante pruebas de laboratorio.
Se han notificado casos de fibrosis sistémica nefrogénica (FSN) asociada al uso de Optimark y de algunos medios de contraste que contienen gadolinio en pacientes con insuficiencia renal grave aguda o crónica (TFG < 30 ml/min/1,73 m2) y/o lesión renal aguda. Optimark está contraindicado en estos pacientes (ver sección 4.3). Los pacientes que se han sometido o se están sometiendo a trasplante hepático están especialmente en situación de riesgo, ya que hay una elevada incidencia de insuficiencia renal aguda en este grupo. Por lo tanto, Optimark no debe utilizarse en pacientes que se hayan sometido o se estén sometiendo a trasplante hepático ni en recién nacidos (ver sección 4.3).
Se desconoce el riesgo de desarrollo de FSN en pacientes con insuficiencia renal moderada (TFG
30-59 ml/min/1,73 m2); por lo tanto, Optimark sólo debe utilizarse tras realizar una meticulosa evaluación de los riesgos-beneficios en pacientes con insuficiencia renal moderada.
La gadoversetamida es dializable. La hemodiálisis poco tiempo después de la administración de Optimark puede ser útil para la eliminación de Optimark del organismo. No existen evidencias que apoyen iniciar hemodiálisis para la prevención o el tratamiento de la FSN en pacientes que no estén en hemodiálisis previamente.
En pacientes con insuficiencia renal basal se ha producido lesión renal aguda que precisó de diálisis con el uso de Optimark. El riesgo de lesión renal aguda puede aumentar con una dosis aumentada del medio de contraste. Debe administrarse la dosis más baja posible para la adquisición de imágenes adecuadas.
Niños y adolescentes
Optimark no se debe administrar con un autoinyector. La dosis necesaria debe ser administrada manualmente a los niños de 2 a 11 años para evitar una sobredosis por error.
Neonatos y lactantes
Optimark no está recomendado para uso en niños menores de dos años de edad debido a la ausencia de datos sobre seguridad y eficacia.
Pacientes de edad avanzada
Dado que el aclaramiento renal de gadoversetamida puede estar deteriorado en los pacientes de edad avanzada, es especialmente importante evaluar a los pacientes de al menos 65 años de edad para descartar la presencia de disfunción renal.
Sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis de hasta 17 ml; esto es, esencialmente "exento de sodio".
Las jeringas de 10 ml y de 15 ml contienen menos de 1 mmol de sodio; esto es, esencialmente
"exentas de sodio".
Dosis mayores contienen 1 mmol de sodio o más, lo que debe ser tenido en cuenta en pacientes con dietas pobres en sodio.
Jeringa precargada
20 ml de solución contienen 28,75 mg de sodio.
30 ml de solución contienen 43,13 mg de sodio.
Vial
20 ml de solución contienen 28,75 mg de sodio.
Niveles plasmáticos de hierro y zinc
En ensayos clínicos se han observado disminuciones transitorias de los niveles plasmáticos de hierro y zinc, por lo que se debe actuar con precaución. Se desconoce la relevancia clínica de estos hallazgos.
4.5 - Interacciones con otros medicamentos de OPTIMARK 500 mcmol/ml Sol. iny. en vial
No se han realizado estudios formales de interacciones.
Se ha demostrado que Optimark causa interferencia en la determinación de los niveles séricos de calcio utilizando el método colorimétrico de la ortocresolftaleína complexona (OCP). Sin embargo, la administración de gadoversetamida no causa una disminución real de los niveles séricos de calcio. En presencia de gadoversetamida, la técnica OCP produce un valor erróneo inferior al nivel sérico real de calcio. La magnitud de este artefacto en la determinación es proporcional a la concentración de gadoversetamida en sangre, y en pacientes con excreción renal normal se pueden obtener valores precisos aproximadamente a los 90 minutos después de la inyección. En pacientes con insuficiencia renal, la eliminación de gadoversetamida será más lenta y la interferencia con la determinación de los niveles séricos de calcio mediante OCP más prolongada. La gadoversetamida no afecta a otros métodos de determinación de los niveles séricos de calcio, como el método colorimétrico arsenazo III, la espectrometría de absorción atómica y la espectrometría de masas con plasma acoplado inductivamente.
4.6 - Embarazo y Lactancia de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Embarazo
No hay datos relativos al uso de gadoversetamida en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3).
No debe utilizarse Optimark durante el embarazo a no ser que la situación clínica de la mujer requiera el uso de gadoversetamida.
Lactancia
Se desconoce si la gadoversetamida se excreta en la leche materna. No se dispone de información suficiente relativa a la excreción de gadoversetamida en la leche de animales. No se puede excluir el riesgo en niños lactantes. Debe interrumpirse la lactancia durante al menos 24 horas después de la administración de Optimark.
Fertilidad
Los datos de los estudios no clínicos no mostraron riesgos especiales para los seres humanos según los estudios convencionales de toxicidad reproductiva. No se han realizado estudios clínicos sobre fertilidad.
4.7 - Efectos sobre la capacidad de conducción de OPTIMARK 500 mcmol/ml Sol. iny. en vial
La influencia de Optimark sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
Los pacientes ambulatorios deben tener en cuenta que, de forma infrecuente (≥1/1.000 a <1/100), pueden ocurrir mareos agudos al conducir vehículos o utilizar máquinas (ver sección 4.8).
4.8 - Reacciones Adversas de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Resumen del perfil de seguridad
La mayoría de las reacciones adversas fueron de intensidad leve a moderada y de naturaleza pasajera. Las reacciones adversas más frecuentes fueron disgeusia, sensación de calor, cefalea y mareo.
Se ha hallado que la mayor parte de las reacciones adversas observadas tras la utilización de gadoversetamida afectaron al sistema nervioso, y, en orden decreciente de frecuencia reacciones adversas generales, trastornos gastrointestinales/ trastornos de la piel y del tejido subcutáneo.
Se han notificado reacciones adversas graves que incluyen reacciones anafilácticas, reacciones cardiovasculares, y trastornos respiratorios alérgicos. El tratamiento debe ser sintomático y debe estar disponible un acceso inmediato a los medicamentos y equipo de emergencia necesarios por si
ocurriese una reacción grave.
Tabla de reacciones adversas
Se han notificado las siguientes reacciones adversas en ensayos clínicos y a partir de la utilización post-comercialización de la gadoversetamida. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
| Sistema de clasificación de órganos (MedDRA) | Frecuentes(≥1/100 a <1/10) | Poco frecuentes (≥1/1.000 a <1/100) | Raras (≥1/10.000 a <1/1.000) | Muy raras (<1/10.000) | No conocida |
| Trastornos del sistema inmunológico | Reacción anafiláctica | ||||
| Trastornos del metabolismo y de la nutrición | Disminución del apetito | ||||
| Trastornos psiquiátricos | Ansiedad, trastornos del sueño, confusión y desorientación | ||||
| Trastornos del sistema nervioso | Cefalea, disgeusia | Mareos, hipoestesia, parestesias, parosmia | Convulsión, temblor, somnolencia, escozor | Síncope | |
| Trastornos oculares | Eritema de párpados, dolor ocular, visión borrosa, conjuntivitis, hiperemia ocular | ||||
| Trastornos del oído y del laberinto | Tinnitus, vértigo | ||||
| Trastornos cardiacos | Palpitaciones, bloqueo A-V de primer grado, extrasístoles, taquicardia, arritmia | ||||
| Trastornos vasculares | Sofocos/rubor | Hipotensión, hipertensión | |||
| Trastornos respiratorios, torácicos y mediastínicos | Congestión nasal, irritación de garganta | Disnea, disfonía, rinorrea, opresión en la garganta, broncoespasmo, tos, edema laríngeo/faríngeo, faringitis, rinitis, estornudos |
| Sistema de clasificación de órganos (MedDRA) | Frecuentes(≥1/100 a <1/10) | Poco frecuentes (≥1/1.000 a <1/100) | Raras (≥1/10.000 a <1/1.000) | Muy raras (<1/10.000) | No conocida |
| Trastornos gastrointestinales | Náuseas, diarrea | Hipersecreción salival, dolor abdominal, estreñimiento, sequedad en la boca | Vómitos | ||
| Trastornos de la piel y del tejido subcutáneo | Prurito, rash | Urticaria, sudor frío, eritema, hiperhidrosis | Edema periorbital | Fibrosis sistémica nefrogénica (FSN) | |
| Trastornos renales y urinarios | Aumento de creatinina en sangre, hematuria | ||||
| Trastornos generales y alteraciones en el lugar de administración | Sensación de calor | Malestar en el pecho, dolor en el pecho, sensación de frío (incluyendo sensación de frío periférica), reacciones en el lugar de administración | Escalofríos, dolor, edema facial, enfermedades asténicas, incluyendo astenia, fatiga y malestar, fiebre, edema periférico, sensaciones extrañas | ||
| Exploraciones complementarias | Calcio en sangre anormal | ALT aumentada, análisis de orina anormal, electrolitos de orina anormales, albúmina en orina, CPK aumentada, hemoglobina reducida | Intervalo QT del electrocardiograma prolongado |
Se han producido reacciones locales en el lugar de inyección y pueden conducir a reacciones de tipo irritación local.
Se han notificado casos de fibrosis sistémica nefrogénica (FSN) con Optimark (ver sección 4.4). Se han notificado casos de placas cutáneas asociadas al gadolinio, con demostración de cuerpos escleróticos en el examen histológico, con algunos medios de contraste que contienen gadolinio en pacientes que, por lo demás, no presentan síntomas ni signos de fibrosis sistémica nefrogénica.
Población pediátrica
Optimark se ha estudiado en niños de 2 años y mayores, observándose un perfil de seguridad similar al de la población adulta.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 - Sobredosificación de OPTIMARK 500 mcmol/ml Sol. iny. en vial
La gadoversetamida se ha ensayado en humanos en dosis de hasta 700 micromoles/kg (siete veces la dosis estándar). No se han descrito las consecuencias clínicas de una sobredosis. Es improbable que se produzcan síntomas de toxicidad aguda en pacientes con una función renal normal. Optimark se puede eliminar mediante hemodiálisis. Sin embargo, no existen evidencias que indiquen que la hemodiálisis sea adecuada para la prevención de la fibrosis sistémica nefrogénica (FSN).
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Grupo farmacoterapéutico: Medios de contraste para RM, código ATC: V08CA06
La gadoversetamida es un quelato que contiene gadolinio -que tiene propiedades paramagnéticas y es responsable del realce del contraste en RM -y el ligando versetamida.
El objetivo de un medio de contraste RM es inducir cambios de intensidad de señal dentro de la lesión facilitando así su diferenciación de las estructuras normales circundantes. Por lo tanto, el uso de un medio de contraste por lo tanto reduce el umbral de detección y visualización de la lesión. Los medios de contraste RM que contienen gadolinio (quelatos de gadolinio) se han diseñado para actuar indirectamente sobre el campo magnético local, alterando los tiempos de relajación protónica T1 (espín-red) y T2 (espín-espín) y a la concentración habitual de 100 micromoles/kg, predomina el acortamiento en T1 y el acortamiento en T2 no es significativo utilizando secuencias ponderadas en T1.
Tras la administración intravenosa, la gadoversetamida, un quelato de gadolinio extracelular, tras la administración intravenosa, se equilibra rápidamente dentro del fluido/espacio extracelular y se elimina principalmente mediante filtración glomerular.
Como resultado de estas características, el momento de la adquisición de la imagen después de la administración del contraste es crítico en el caso de imágenes hepáticas. Para las metástasis hepáticas, la diferencia de señal entre el tumor y el tejido hepático circundante aumenta significativamente durante los primeros 90 segundos después de administrar un medio de contraste de gadolinio extracelular. Por lo tanto, debe iniciarse una secuencia rápida de imagen 20 segundos después de la inyección en bolo del medio de contraste, cuando el medio se localiza predominantemente en las arterias hepáticas y a continuación de nuevo a los 60 segundos después de la inyección durante la fase venosa portal dominante. Puesto que los sistemas arterial y venoso portal hepáticos suministran aproximadamente el 20 % y el 80 % del aporte sanguíneo a nivel hepático, respectivamente, las
imágenes precoces (fase arterial hepática) proporcionan una óptima visualización de la lesión en el
caso de lesiones hipervasculares y las imágenes adquiridas durante la fase venosa portal son útiles para lesiones hipovasculares (la mayoría de las lesiones metastásicas son relativamente hipovasculares y se visualizan mejor durante la fase venosa portal, manifestándose como áreas de menor intensidad de señal en comparación con el hígado, con gran intensidad de señal. La visibilidad de las lesiones hipo e hípervasculares puede quedar reducida si la adquisición de la imagen se demora más de tres minutos debido a la difusión del medio de contraste en los espacios intersticiales tanto del parénquima hepático como de la lesión (por ejemplo metástasis) haciendo que la lesión sea isointensa respecto al
parénquima hepático normal. Las imágenes tardías tras la administración del contraste o imágenes en fase de equilibrio (obtenidas más de 5 minutos tras la administración del medio de contraste) ayudan a la caracterización de lesiones, por ejemplo el centro de una metástasis puede acumular medio de contraste en el espacio intersticial de la lesión y volverse hiperintenso respecto al hígado normal. Esta diferencia en el patrón de intensificación es útil para realizar un diagnóstico diferencial basado en la caracterización de la lesión y en la confianza diagnóstica.
El realce del contraste en los tumores cerebrales utilizando un medio de contraste que contenga gadolinio (o yodo) depende de la integridad de la barrera hematoencefálica (BHE). Por ello, estos medios de contraste se han denominado marcadores localizaciones con alteración de la BHE. Cuando se interrumpe la continuidad de la BHE, las moléculas de gadoversetamida difunden en el compartimiento intersticial produciendo así el efecto paramagnético característico de acortamiento de T1 y T2. En general, la adición de contraste a RM, a la dosis clínica estándar de 100 micromoles/kg, mejoró significativamente la detección de las lesiones, la sensibilidad y precisión diagnóstica.
5.2 - Propiedades farmacocinéticas de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Distribución
La farmacocinética de la gadoversetamida se ajusta a un modelo abierto bicompartimental. A la dosis de 100 micromol/kg, la semivida media de distribución en sujetos sanos calculada por el método de los residuales en 12 voluntarios sanos es 13,3 ± 6,8 min. El volumen de distribución medio a la dosis de 100 micromol/kg en pacientes sin insuficiencia renal (incluyendo tanto sujetos sanos como pacientes con patología de SNC o hepática) fue 158,7 ± 29,0 a 214,3 (intervalo 116,4 a 295,0) ml/kg. Este volumen de distribución (aproximadamente 10-15 l para un peso corporal de 70 kg) es coherente
con medicamentos que se distribuyen en el fluido extracelular. La dosis no afectó de forma consistente al volumen de distribución en ninguno de los ensayos. La gadoversetamida no presenta unión a proteínas in vitro.
Eliminación
La semivida de eliminación a la dosis de 100 micromol/kg varía desde 1,49 ± 0,15 h en voluntarios sanos hasta 2,11 ± 0,62 h en pacientes sin insuficiencia renal (incluyendo sujetos sanos como pacientes con patología de SNC o hepática).
El aclaramiento plasmático medio de gadoversetamida en sujetos sanos (111,0 ± 14,1 ml/min/1,73 m²) no difiere significativamente del aclaramiento renal medio. Se obtienen resultados similares en sujetos normales y pacientes con diversas combinaciones de insuficiencia hepática, del SNC y renal, y el aclaramiento renal de gadoversetamida supone aproximadamente el 95 % del aclaramiento plasmático total. Estos resultados (relación aclaramiento renal/plasmático total cercana a 1) indican que la gadoversetamida se elimina principalmente a través de los riñones.
No hubo ninguna diferencia sistemática en ninguno de los parámetros cinéticos en relación con la dosis (100 a 700 micromol/kg). Por lo tanto, dentro de este rango de dosis, la cinética de la gadoversetamida es lineal.
Metabolismo
La mayor parte de la dosis se encuentra en forma de complejo intacto en la orina, lo que sugiere que no se metaboliza significativamente de la gadoversetamida en seres humanos.
Poblaciones especiales
Influencia del género:
Sujetos adultos de sexo masculino y femenino participaron en dos ensayos farmacocinéticos. No se identificaron diferencias significativas en la farmacocinética dependientes del género.
Efectos de la edad:
Al ajustar según el peso corporal, el aclaramiento total de gadoversetamida es mayor en el grupo de edad de 2 a 11 años (143 ± 27,9 ml/h/kg) que el observado en el grupo de edad de 12 a 18 años (117 ±
26,1 ml/h/kg) y las dos poblaciones adultas (82,1 ± 16,8 y 56,5 ± 9,7 ml/h/kg en los grupos de edad de
19 a 64 y ≥ 65 años, respectivamente).
La semivida de eliminación en los grupos de edad de 2 a 11 y 12 a 18 años (1,4 ± 0,3 y 1,6 ± 0,3 h-1, respectivamente) es más corta que la observada en las dos poblaciones adultas (1,9 ± 0,5 y 2,5 ± 0,5 h-
1 en los grupos de edad de 19 a 64 y ≥ 65 años, respectivamente). El número de pacientes de edad
avanzada en los que se caracterizó la farmacocinética fue limitado (más de 65 años, N=3).
Efecto de la insuficiencia renal
Los niveles plasmáticos de gadoversetamida aumentan linealmente cuando la función renal disminuye;
en pacientes con insuficiencia renal grave (CrCl<30 ml/min) ), lo que produce incluso a una eliminación seis veces menor de gadoversetamida y a un consiguiente aumento de seis veces de la magnitud de la exposición, según AUC y t½. La gadoversetamida se administra solamente como dosis única lo que limita la duración de esta exposición más intensa y prolongada. Con todo, casi la totalidad de la dosis se recupera en la orina después de 72 horas incluso en pacientes con insuficiencia renal grave y en voluntarios sanos se administraron dosis de hasta 500 micromol/kg sin problemas de seguridad. Sin embargo, dado que se han notificado casos de FSN que podrían estar asociados con la
insuficiencia renal tras el uso de gadoversetamida y otros medios de contraste que contienen gadolinio, Optimark no debe utilizarse en estos pacientes.
5.3 - Datos preclínicos sobre seguridad de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad aguda, toxicidad reproductiva, tolerancia local, antigenicidad y genotoxicidad. No se han realizado estudios de carcinogenicidad.
Estudios de toxicidad a dosis repetidas en ratas y perros revelaron la formación de vacuolas en las células tubulares de los riñones, con una fuerte evidencia que apunta a la reversibilidad de este efecto. No se observaron daños funcionales.
La eliminación de Optimark en perros menores de 3 meses de edad presentó un retraso significativo debido a la inmadurez de la función renal y provocó una elevada exposición sistémica a Optimark. La dosificación repetida semanalmente de dos a veinte veces la dosis clínica a partir de la semana de edad hasta la maduración generó una extensa mineralización tisular que produjo efectos localizados, como dermatitis ulcerosa, compromiso de la circulación y disfunción hepática.
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Versetamida Hidróxido de calcio Cloruro de calcio dihidrato
Hidróxido de sodio y/o ácido clorhídrico para ajuste del pH. Agua para preparaciones inyectables.
6.2 - Incompatibilidades de OPTIMARK 500 mcmol/ml Sol. iny. en vial
En ausencia de estudios de compatibilidad, Optimark no debe mezclarse con otros medicamentos.
6.3 - Período de validez de OPTIMARK 500 mcmol/ml Sol. iny. en vial
3 años.
Se ha demostrado estabilidad en-uso química y física durante 24 horas a temperaturas de hasta 25°C. Por razones de seguridad microbiológica, el producto debe utilizarse inmediatamente. Si no se utiliza inmediatamente, la responsabilidad sobre el tiempo de almacenamiento en uso y las condiciones previas a la utilización recae sobre el usuario.
6.4 - Precauciones especiales de conservación de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Jeringa precargada
Conservar la jeringa en el embalaje exterior para protegerla de la luz.
Vial
Conservar el vial en el embalaje exterior para protegerlo de la luz.
No refrigerar o congelar.
Para las condiciones de conservación tras la primera apertura del medicamento, ver sección 6.3.
6.5 - Naturaleza y contenido del recipiente de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Jeringa precargada
Optimark se presenta en jeringas precargadas hechas de polipropileno. La cápsula de la punta de la jeringa y el émbolo están hechos de goma de bromobutilo.
Tamaños de envases:
1 x 10 ml 10 x 10 ml
1 x 15 ml 10 x 15 ml
1 x 20 ml 10 x 20 ml
1 x 30 ml 10 x 30 ml
Puede que solamente estén comercializados algunos tamaños de envases. Vial
Optimark se presenta en viales de vidrio de borosilicato incoloro de alta resistencia (EP Tipo I). Los viales llevan tapones de goma de bromobutilo, cápsulas de cierre de aluminio, y tapas de tipo "flip- off".
Tamaños de envases:
1 x 10 ml 10 x 10 ml
1 x 15 ml 10 x 15 ml
1 x 20 ml 10 x 20 ml
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de OPTIMARK 500 mcmol/ml Sol. iny. en vial
Optimark está destinado a un solo uso; la solución no utilizada debe desecharse.
La solución no debe utilizarse en caso de que presente alteraciones del color o si aparecen partículas. Si se utiliza equipamiento no desechable, se debe prestar una escrupulosa atención para impedir cualquier contaminación residual con trazas de medios de limpieza.
Jeringa precargada
Jeringas precargadas
Inspección y montaje
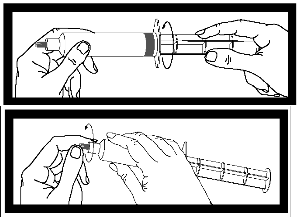
Inspeccione la jeringa para detectar posibles signos de escape. No la utilice si observa un escape.
| Tras enroscar la varilla en el émbolo de la jeringa, es importante girar la varilla media vuelta más, de modo que el émbolo gris rote libremente | |
| Antes de usar la jeringa, desenrosque la cápsula gris de la punta y deséchela. La jeringa está ahora lista para acoplarla a la aguja o a la vía de perfusión. |
Elimine la jeringa y la solución sobrante después del uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con las normativas locales.
La etiqueta de seguimiento despegable de las jeringas precargadas debe adherirse a la historia del paciente para permitir llevar un registro exacto del medio de contraste de gadolinio utilizado. También debe registrarse la dosis utilizada.
Si se utilizan historias del paciente electrónicas, el nombre del medicamento, el número de lote y la dosis se deben introducir en la historia del paciente.
Vial
Optimark sólo debe extraerse con la jeringa inmediatamente antes de ser usado.
El producto debe ser examinado antes de su utilización para confirmar que todos los sólidos estén disueltos y que el envase y el cierre no estén dañados. Si quedan sólidos, el vial debe eliminarse.
Elimine la jeringa y la solución sobrante después del uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con las normativas locales.
La etiqueta de seguimiento despegable de los viales debe adherirse a la historia del paciente para permitir llevar un registro exacto del medio de contraste de gadolinio utilizado. También debe registrarse la dosis utilizada. Si se utilizan historias del paciente electrónicas, el nombre del medicamento, el número de lote y la dosis se deben introducir en la historia del paciente.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Guerbet
15, rue des Vanesses
93420 Villepinte
Francia
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Jeringa precargada
1 x 10 ml: EU/1/07/398/007
10 x 10 ml: EU/1/07/398/008
1 x 15 ml: EU/1/07/398/009
10 x 15 ml: EU/1/07/398/010
1 x 20 ml: EU/1/07/398/011
10 x 20 ml: EU/1/07/398/012
1 x 30 ml: EU/1/07/398/013
10 x 30 ml EU/1/07/398/014
Vial
1 x 10 ml: EU/1/07/398/001
10 x 10 ml: EU/1/07/398/002
1 x 15 ml: EU/1/07/398/003
10 x 15 ml: EU/1/07/398/004
1 x 20 ml: EU/1/07/398/005
10 x 20 ml: EU/1/07/398/006
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 23 de julio de 2007
Fecha de la última renovación: 15 de junio de 2012
10. - FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia
Europea de Medicamentos http://www.ema.europa.eu/.

