SYNJARDY 12,5 MG/1000 MG COMPRIMIDOS RECUBIERTOS CON PELICULA

| ATC: Metformina y empagliflozina |
| PA: Metformina hidrocloruro, Empagliflozina |
Envases
- Env. con 60
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica Aportación reducida por el beneficiario
Aportación reducida por el beneficiario- Fi: Medicamento incluido en la financiación del SNS
- Facturable SNS: SI
- Comercializado: Si
- Situación: Alta
- Código Nacional: 706803
- EAN13: 8470007068037
- Conservar en frío: No
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Synjardy 5 mg/850 mg comprimidos recubiertos con película
Cada comprimido contiene 5 mg de empagliflozina y 850 mg de hidrocloruro de metformina.
Synjardy 5 mg/1.000 mg comprimidos recubiertos con película
Cada comprimido contiene 5 mg de empagliflozina y 1 000 mg de hidrocloruro de metformina.
Synjardy 12,5 mg/850 mg comprimidos recubiertos con película
Cada comprimido contiene 12,5 mg de empagliflozina y 850 mg de hidrocloruro de metformina.
Synjardy 12,5 mg/1.000 mg comprimidos recubiertos con película
Cada comprimido contiene 12,5 mg de empagliflozina y 1 000 mg de hidrocloruro de metformina.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. - FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Synjardy 5 mg/850 mg comprimidos recubiertos con película
Comprimidos recubiertos con película de color blanco amarillento, ovalados, biconvexos, grabados con la inscripción «S5» y el logotipo de Boehringer Ingelheim en una cara y la inscripción «850» en la otra (longitud del comprimido: 19,2 mm, anchura del comprimido: 9,4 mm).
Synjardy 5 mg/1.000 mg comprimidos recubiertos con película
Comprimidos recubiertos con película de color amarillo pardusco, ovalados, biconvexos, grabados con la inscripción «S5» y el logotipo de Boehringer Ingelheim en una cara y la inscripción «1 000» en la otra (longitud del comprimido: 21,1 mm, anchura del comprimido: 9,7 mm).
Synjardy 12,5 mg/850 mg comprimidos recubiertos con película
Comprimidos recubiertos con película de color blanco rosado, ovalados, biconvexos, grabados con la inscripción «S12» y el logotipo de Boehringer Ingelheim en una cara y la inscripción «850» en la otra (longitud del comprimido: 19,2 mm, anchura del comprimido: 9,4 mm).
Synjardy 12,5 mg/1.000 mg comprimidos recubiertos con película
Comprimidos recubiertos con película de color morado pardusco oscuro, ovalados, biconvexos, grabados con la inscripción «S12» y el logotipo de Boehringer Ingelheim en una cara y la inscripción «1 000» en la otra (longitud del comprimido: 21,1 mm, anchura del comprimido: 9,7 mm).
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Synjardy está indicado en adultos y niños a partir de 10 años de edad para el tratamiento de la diabetes mellitus tipo 2 como tratamiento asociado a dieta y ejercicio:
- en pacientes no suficientemente controlados con su dosis máxima tolerada de metformina sola
- en combinación con otros medicamentos, para el tratamiento de la diabetes en pacientes no suficientemente controlados con metformina y estos medicamentos
- en pacientes que ya se estén tratando con la combinación de empagliflozina y metformina en comprimidos separados.
Para consultar los resultados de los ensayos respecto a las combinaciones, los efectos en el control glucémico y los acontecimientos cardiovasculares, así como las poblaciones estudiadas, ver las secciones 4.4, 4.5 y 5.1.
4.2 - Posología y administración de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
4.3 - Contraindicaciones de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
- Hipersensibilidad a los principios activos o a alguno de los excipientes incluidos en la sección 6.1.
- Cualquier tipo de acidosis metabólica aguda (como acidosis láctica, cetoacidosis diabética) (ver sección 4.4).
- Precoma diabético.
- Insuficiencia renal grave (TFGe< 30 ml/min/1,73 m2) (ver las secciones 4.2 y 4.4).
- Cuadros agudos que puedan alterar la función renal, como por ejemplo: deshidratación, infección grave o shock (ver las secciones 4.4 y 4.8).
- Enfermedad que pueda producir hipoxia tisular (especialmente enfermedad aguda, o empeoramiento de enfermedad crónica), como por ejemplo: insuficiencia cardíaca descompensada, insuficiencia respiratoria, infarto de miocardio reciente, shock (ver sección 4.4).
- Insuficiencia hepática, intoxicación etílica aguda, alcoholismo (ver las secciones 4.2 y 4.5).
4.4 - Advertencias y Precauciones de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Generales
Empagliflozina no se debe usar en pacientes con diabetes mellitus tipo 1 (ver “Cetoacidosis diabética” en la sección 4.4).
Acidosis láctica
La acidosis láctica es una complicación metabólica muy rara, pero grave, que se produce con mayor frecuencia durante el empeoramiento agudo de la función renal o en caso de enfermedad cardiorrespiratoria o septicemia. La acumulación de metformina se produce durante el empeoramiento agudo de la función renal e incrementa el riesgo de acidosis láctica.
En caso de deshidratación (diarrea o vómitos intensos, fiebre o reducción de la ingesta de líquidos), se debe interrumpir metformina de forma temporal y se recomienda contactar con un profesional sanitario.
Los medicamentos que puedan alterar de manera aguda la función renal (como antihipertensivos, diuréticos y AINE) se deben iniciar con precaución en los pacientes tratados con metformina. Otros factores de riesgo para la acidosis láctica son el consumo excesivo de alcohol, la insuficiencia hepática, la diabetes mal controlada, la cetosis, el ayuno prolongado y cualquier proceso asociado a hipoxia, así como el uso concomitante de medicamentos que puedan causar acidosis láctica (ver las secciones 4.3 y 4.5).
Se debe informar a los pacientes o a los cuidadores acerca del riesgo de acidosis láctica. La acidosis láctica se caracteriza por disnea acidótica, dolor abdominal, calambres musculares, astenia e hipotermia, seguidos de coma. En caso de que se sospeche la presencia de síntomas, el paciente debe dejar de tomar metformina y buscar atención médica inmediata. Los hallazgos diagnósticos de laboratorio son una disminución del pH sanguíneo (< 7,35), niveles de lactato plasmático aumentados (> 5 mmol/l) y un aumento del desequilibrio aniónico y del cociente lactato/piruvato.
Cetoacidosis diabética
Se han notificado casos raros de cetoacidosis diabética (CAD), incluidos casos potencialmente mortales y con desenlace mortal, en pacientes tratados con inhibidores del SGLT2, incluida empagliflozina. En algunos de estos casos, la presentación del cuadro clínico fue atípica, con un ascenso moderado de los niveles de glucosa en sangre, por debajo de 14 mmol/l (250 mg/dl). Se desconoce si la CAD puede ocurrir con mayor probabilidad con dosis mayores de empagliflozina.
El riesgo de cetoacidosis diabética se debe considerar en caso de síntomas inespecíficos como náuseas, vómitos, anorexia, dolor abdominal, sed excesiva, dificultad para respirar, confusión, fatiga o somnolencia inusuales. Se debe evaluar a los pacientes de forma inmediata para detectar la cetoacidosis en caso que aparezcan estos síntomas, independientemente del nivel de glucosa en sangre.
En pacientes en los que se sospeche o diagnostique CAD, el tratamiento con empagliflozina se debe suspender inmediatamente.
Se debe interrumpir el tratamiento en pacientes que están hospitalizados por un procedimiento quirúrgico mayor o enfermedades agudas graves. Se recomienda controlar las cetonas en estos pacientes. Se prefiere la determinación de los niveles de cuerpos cetónicos en sangre a la determinación en orina. El tratamiento con empagliflozina se puede reanudar cuando los valores de cuerpos cetónicos sean normales y el estado del paciente se haya estabilizado.
Antes de iniciar empagliflozina, se deben considerar los antecedentes del paciente que puedan predisponer a cetoacidosis.
Los pacientes que pueden tener un riesgo mayor de CAD son aquellos pacientes con baja reserva de células beta funcionales (p. ej., pacientes con diabetes tipo 2 con péptido C bajo o con diabetes autoinmune latente del adulto (LADA) o pacientes con antecedentes de pancreatitis), pacientes con trastornos que den lugar a una ingesta restringida de alimentos o a una deshidratación grave, pacientes cuyas dosis de insulina estén reducidas y pacientes con mayores necesidades de insulina debido a una enfermedad médica aguda, cirugía o abuso de alcohol. Los inhibidores del SGLT2 se deben usar con precaución en estos pacientes.
No se recomienda reiniciar el tratamiento con un inhibidor del SGLT2 en pacientes con CAD mientras estaban en tratamiento con un inhibidor del SGLT2, a menos que se haya identificado y resuelto claramente otro factor desencadenante.
Synjardy no se debe utilizar en pacientes con diabetes tipo 1. Los datos de un programa de ensayos clínicos en pacientes con diabetes tipo 1 mostraron un aumento de la incidencia de CAD con una frecuencia frecuente en pacientes tratados con 10 mg y 25 mg de empagliflozina como tratamiento complementario de la insulina en comparación con un placebo.
Administración de medios de contraste yodados
La administración intravascular de medios de contraste yodados puede provocar nefropatía inducida por el contraste, que puede ocasionar la acumulación de metformina y puede aumentar el riesgo de acidosis láctica. Por tanto, la administración de metformina se debe interrumpir antes o en el momento de la prueba y no se debe reanudar hasta pasadas al menos 48 horas, siempre que se haya reevaluado la función renal y comprobado que es estable (ver las secciones 4.2 y 4.5).
Insuficiencia renal
Debido a su mecanismo de acción, una disminución de la función renal provocará una reducción de la eficacia glucémica de empagliflozina. El tratamiento con empagliflozina/metformina está contraindicado en pacientes con TFGe < 30 ml/min/1,73 m2 y se debe interrumpir de forma temporal en presencia de trastornos que alteren la función renal (ver sección 4.3).
Monitorización de la función renal
Se recomienda evaluar la función renal tal como se indica a continuación:
- antes de iniciar el tratamiento con empagliflozina/metformina y periódicamente durante el tratamiento, al menos una vez al año (ver sección 4.2).
- antes de iniciar el tratamiento con cualquier medicamento concomitante que pueda tener un impacto negativo en la función renal.
Función cardíaca
Los pacientes con insuficiencia cardíaca tienen un riesgo mayor de hipoxia y de insuficiencia renal. En pacientes con insuficiencia cardíaca crónica estable se puede usar Synjardy con una monitorización regular de la función cardíaca y renal. Synjardy está contraindicado en los pacientes con insuficiencia cardíaca aguda e inestable debido al componente metformina (ver sección 4.3).
Cirugía
Metformina se debe suspender en el momento de la cirugía con anestesia general, espinal o epidural. El tratamiento se puede reanudar pasadas 48 horas desde la cirugía o tras la reanudación de la nutrición oral, siempre que se haya reevaluado la función renal y comprobado que es estable.
Riesgo de hipovolemia
En base al modo de acción de los inhibidores del SGLT2, la diuresis osmótica que acompaña a la glucosuria terapéutica puede provocar una disminución moderada de la presión arterial (ver sección 5.1). Por lo tanto, se debe tener precaución en los pacientes para los que una caída de la presión arterial inducida por empagliflozina pudiera suponer un riesgo, tales como pacientes con enfermedad cardiovascular conocida, pacientes en tratamiento antihipertensivo con antecedentes de hipotensión o pacientes de 75 años de edad o mayores.
En caso de enfermedades que puedan conducir a una pérdida de líquidos (por ejemplo, una enfermedad gastrointestinal), se recomienda una estrecha supervisión de la volemia (por ejemplo, exploración física, medición de la presión arterial, pruebas de laboratorio, incluyendo el hematocrito) y de los electrolitos en pacientes que reciben Synjardy. Se debe valorar la interrupción temporal del tratamiento con Synjardy hasta que se corrija la pérdida de líquidos.
Pacientes de edad avanzada
El efecto de empagliflozina en la eliminación de glucosa por la orina se asocia a la diuresis osmótica, lo que podría afectar al estado de hidratación. Los pacientes de 75 años de edad o mayores pueden presentar un mayor riesgo de hipovolemia. Por tanto, se debe prestar especial atención a la ingesta de líquidos en caso de que se administre de forma conjunta con medicamentos que puedan producir hipovolemia (p. ej., diuréticos, inhibidores de la ECA).
Infecciones del tracto urinario
Durante el periodo poscomercialización se han notificado casos de infecciones complicadas del tracto urinario incluyendo pielonefritis y urosepsis en pacientes tratados con empagliflozina (ver sección 4.8). En el caso de pacientes con infecciones complicadas del tracto urinario, se debe valorar la interrupción temporal del tratamiento.
Fascitis necrosante del perineo (gangrena de Fournier)
Se han notificado casos poscomercialización de fascitis necrosante del perineo (también conocida como gangrena de Fournier) en pacientes de ambos sexos tratados con inhibidores del SGLT2. Se trata de un acontecimiento raro, pero grave y potencialmente mortal, que requiere intervención quirúrgica urgente y tratamiento antibiótico.
Se indicará a los pacientes que acudan al médico si presentan una combinación de síntomas como dolor, dolor a la palpación, eritema o inflamación en la región genital o perineal, con fiebre o malestar general. Tenga en cuenta que la infección urogenital o el absceso perineal pueden preceder a la fascitis necrosante. Si se sospecha gangrena de Fournier, se debe interrumpir Synjardy e instaurar un tratamiento inmediato (incluidos antibióticos y desbridamiento quirúrgico).
Amputaciones de miembros inferiores
Se ha observado un incremento en los casos de amputación de miembros inferiores (principalmente de los dedos de los pies) en ensayos clínicos a largo plazo con otro inhibidor del SGLT2. Se desconoce si esto constituye un efecto de clase. Al igual que para todos los pacientes diabéticos, es importante aconsejar a los pacientes acerca del cuidado rutinario preventivo de los pies.
Lesión hepática
Se han notificado casos de lesión hepática con el uso de empagliflozina en ensayos clínicos. No se ha establecido una relación causal entre empagliflozina y la lesión hepática.
Aumento del hematocrito
Se ha observado un aumento del hematocrito con el tratamiento con empagliflozina (ver sección 4.8).
Enfermedad renal crónica
Se dispone de experiencia con empagliflozina para el tratamiento de la diabetes en pacientes con enfermedad renal crónica (TFGe ≥ 30 ml/min/1,73 m2) con y sin albuminuria. Los pacientes con albuminuria se pueden beneficiar más del tratamiento con empagliflozina.
Análisis de orina
Debido a su mecanismo de acción, los pacientes que están tomando Synjardy presentarán un resultado positivo para la glucosa en la orina.
Interferencia con la prueba del 1,5-anhidroglucitol (1,5-AG)
No se recomienda la monitorización del control de la glucemia con la prueba del 1,5-AG, ya que las mediciones de 1,5-AG no son fiables para valorar el control de la glucemia en pacientes que toman inhibidores del SGLT2. Se recomienda el uso de métodos alternativos para la monitorización del control de la glucemia.
Vitamina B12
Metformina puede reducir los niveles de vitamina B12. El riesgo de niveles bajos de vitamina B12 aumenta al aumentar la dosis de metformina, la duración del tratamiento y/o en pacientes con factores de riesgo que se ha demostrado que causan déficit de vitamina B12. En caso de sospecha de déficit de vitamina B12 (por ejemplo, anemia o neuropatía), se deben vigilar los niveles séricos de vitamina B12. Podría ser necesario un seguimiento periódico de la vitamina B12 en pacientes con factores de riesgo de déficit de vitamina B12. El tratamiento con metformina se debe continuar mientras se tolere y no esté contraindicado y se debe administrar el tratamiento corrector apropiado para el déficit de vitamina B12 conforme a las guías clínicas actuales.
Población pediátrica
En el ensayo DINAMO (ver sección 5.1), el perfil de seguridad general en niños y adolescentes fue similar al perfil de seguridad conocido observado en pacientes adultos, y no se observaron diferencias relevantes entre placebo y empagliflozina en relación con las evaluaciones del crecimiento o con la maduración sexual después de 26 semanas de tratamiento.
No se ha detectado un efecto de metformina en el crecimiento y la pubertad durante estudios clínicos controlados de un año de duración, pero no se dispone de datos a largo plazo sobre estos aspectos específicos. Por consiguiente, se recomienda un seguimiento meticuloso del efecto de metformina en estos parámetros en niños tratados con metformina, especialmente en niños prepúberes.
Niños de entre 10 y 12 años de edad
En estudios de metformina, solo se incluyó a 15 pacientes de entre 10 y 12 años de edad en los estudios clínicos controlados realizados en niños y adolescentes.
En el ensayo DINAMO se incluyó a 157 pacientes, de los cuales el 91 % estaba recibiendo metformina como tratamiento de base; 25 de estos pacientes tenían entre 10 y 12 años de edad. Aunque la eficacia y seguridad de metformina en estos niños no se diferenciaron de la eficacia y seguridad en niños mayores y adolescentes, se recomienda precaución especial al prescribir el medicamento a niños de entre 10 y 12 años de edad.
4.5 - Interacciones con otros medicamentos de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
La administración conjunta de dosis múltiples de empagliflozina y metformina no altera de forma significativa la farmacocinética ni de empagliflozina ni de metformina en individuos sanos.
No se han realizado estudios de interacciones con Synjardy. A continuación se refleja la información disponible sobre los principios activos individuales.
Empagliflozina
Interacciones farmacodinámicas
Diuréticos
Empagliflozina puede aumentar el efecto diurético de las tiazidas y de los diuréticos del asa y puede aumentar el riesgo de deshidratación e hipotensión (ver sección 4.4).
Insulina y secretagogos de insulina
La insulina y los secretagogos de insulina, como las sulfonilureas, pueden aumentar el riesgo de hipoglucemia. Por lo tanto, se puede necesitar una dosis más baja de insulina o de un secretagogo de insulina para disminuir el riesgo de hipoglucemia cuando éstos se usan en combinación con empagliflozina (ver las secciones 4.2 y 4.8).
Interacciones farmacocinéticas
Efectos de otros medicamentos sobre empagliflozina
Los datos in vitro sugieren que la principal vía metabólica de empagliflozina en humanos es la glucuronidación por las uridina 5'-difosfoglucuronosiltransferasas UGT1A3, UGT1A8, UGT1A9 y UGT2B7. Empagliflozina es un sustrato de los transportadores de captación humanos OAT3, OATP1B1, y OATP1B3, pero no de OAT1 y OCT2. Empagliflozina es un sustrato de la glicoproteína-P (gp-P) y la proteína de resistencia al cáncer de mama (BCRP).
La administración conjunta de empagliflozina con probenecid, un inhibidor de las enzimas UGT y del OAT3, dio lugar a un aumento del 26 % en las concentraciones plasmáticas máximas (Cmax) de empagliflozina y a un aumento del 53 % en el área bajo la curva concentración-tiempo (AUC). Estos cambios no se consideraron clínicamente significativos.
No se ha estudiado el efecto de la inducción de la UGT (p. ej., inducción por rifampicina o por fenitoína) sobre empagliflozina. No se recomienda el tratamiento concomitante con inductores de las enzimas UGT debido al riesgo potencial de que disminuya la eficacia. Si se debe administrar de forma concomitante un inductor de estas enzimas UGT, se recomienda vigilar el control de la glucemia para determinar que la respuesta a Synjardy es adecuada.
Un estudio de interacción con gemfibrozilo, un inhibidor in vitro de los transportadores OAT3 y OATP1B1/1B3, mostró que la Cmax de empagliflozina aumentaba en un 15 % y que el AUC aumentaba en un 59 % después de la administración conjunta. Estos cambios no se consideraron clínicamente significativos.
La inhibición de los transportadores OATP1B1/1B3 mediante la administración conjunta de rifampicina dio lugar a un aumento del 75 % en la Cmax y a un aumento del 35 % en el AUC de empagliflozina. Estos cambios no se consideraron clínicamente significativos.
La exposición a empagliflozina fue similar con y sin la administración conjunta de verapamilo, un inhibidor de la gp-P, lo que indica que la inhibición de la gp-P no tiene un efecto clínicamente relevante sobre empagliflozina.
Los estudios de interacción sugieren que la farmacocinética de empagliflozina no se vio influida por la administración conjunta de metformina, glimepirida, pioglitazona, sitagliptina, linagliptina, warfarina, verapamilo, ramipril, simvastatina, torasemida e hidroclorotiazida.
Efectos de empagliflozina sobre otros medicamentos
Empagliflozina puede aumentar la excreción renal de litio y reducir así los niveles sanguíneos de litio. Se debe monitorizar la concentración sérica de litio con mayor frecuencia después del inicio del tratamiento y de los cambios de dosis de empagliflozina. Derive al paciente al médico que le prescribió el litio para que le monitorice la concentración sérica de litio.
De acuerdo a los estudios in vitro, empagliflozina no inhibe, inactiva ni induce las isoformas del CYP450. Empagliflozina no inhibe la UGT1A1, la UGT1A3, la UGT1A8, la UGT1A9 ni la UGT2B7. Por lo tanto, se considera improbable que se produzcan interacciones farmacológicas que impliquen a las principales isoformas del CYP450 y la UGT con empagliflozina y a los sustratos de estas enzimas administrados de forma conjunta.
Empagliflozina no inhibe la gp-P a dosis terapéuticas. De acuerdo a los estudios in vitro, se considera improbable que empagliflozina provoque interacciones con principios activos que sean sustratos de la gp P. La administración conjunta de digoxina, un sustrato de la gp-P, con empagliflozina dio lugar a un aumento del 6 % en el AUC y un aumento del 14 % en la Cmax de digoxina. Estos cambios no se consideraron clínicamente significativos.
Empagliflozina no inhibe in vitro a los transportadores de captación humanos, tales como OAT3, OATP1B1 y OATP1B3 a concentraciones plasmáticas clínicamente relevantes y, como tales, las interacciones farmacológicas con sustratos de estos transportadores de captación se consideran improbables.
Los estudios de interacción realizados en voluntarios sanos sugieren que empagliflozina no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de metformina, glimepirida, pioglitazona, sitagliptina, linagliptina, simvastatina, warfarina, ramipril, digoxina, los diuréticos y los anticonceptivos orales.
Metformina
Uso concomitante no recomendado
Alcohol
La intoxicación alcohólica está asociada con un mayor riesgo de acidosis láctica, especialmente en caso de ayuno, desnutrición o insuficiencia hepática.
Transportadores de cationes orgánicos (OCT, por sus siglas en inglés)
Metformina es un sustrato de los transportadores OCT1 y OCT2. La administración conjunta de metformina con
- Inhibidores del OCT1 (como verapamilo) puede reducir la eficacia de metformina.
- Inductores del OCT1 (como rifampicina) puede aumentar la absorción gastrointestinal y la eficacia de metformina.
- Inhibidores del OCT2 (como cimetidina, dolutegravir, ranolazina, trimetoprima, vandetanib e isavuconazol) puede disminuir la eliminación renal de metformina y, por tanto, dar lugar a un aumento de la concentración plasmática de metformina.
- Inhibidores tanto del OCT1 como del OCT2 (como crizotinib y olaparib) puede alterar la eficacia y la eliminación renal de metformina.
Por tanto, se recomienda tener precaución, especialmente en pacientes con insuficiencia renal, cuando se administren estos medicamentos de forma conjunta con metformina, ya que podría aumentar la concentración plasmática de metformina. En caso necesario, se puede considerar la posibilidad de ajustar la dosis de metformina, ya que los inhibidores/inductores de los OCT pueden alterar la eficacia de metformina (ver las secciones 4.2 y 4.4).
Medios de contraste yodados
La administración de metformina se debe interrumpir antes o en el momento de la prueba y no se debe reanudar hasta pasadas al menos 48 horas, siempre que se haya reevaluado la función renal y comprobado que es estable (ver las secciones 4.2 y 4.4).
Combinaciones que requieren precauciones de empleo
Algunos medicamentos pueden afectar de forma adversa la función renal, lo que puede incrementar el riesgo de acidosis láctica, p. ej., los AINEs, incluidos los inhibidores selectivos de la ciclooxigenasa (COX) II, los inhibidores de la ECA, los antagonistas del receptor de la angiotensina II y los diuréticos, en especial, los diuréticos del asa. Cuando se inicien o se utilicen estos productos en combinación con metformina, es necesario supervisar de manera estrecha la función renal.
Los glucocorticoides (administrados por vía sistémica y local), los agonistas beta-2 y los diuréticos poseen actividad hiperglucémica intrínseca. Se debe informar al paciente y se deben realizar controles de glucosa en sangre más frecuentes, especialmente al inicio del tratamiento con este tipo de medicamentos. En caso necesario, se debe ajustar la dosis del medicamento antihiperglucémico durante el tratamiento con el otro medicamento y cuando éste se suspenda.
Insulina y secretagogos de insulina
La insulina y los secretagogos de insulina, como las sulfonilureas, pueden aumentar el riesgo de hipoglucemia. Por lo tanto, se puede necesitar una dosis más baja de insulina o de un secretagogo de insulina para disminuir el riesgo de hipoglucemia cuando éstos se usan en combinación con metformina (ver las secciones 4.2 y 4.8).
Población pediátrica
Solo se han realizado estudios de interacciones en adultos.
4.6 - Embarazo y Lactancia de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Embarazo
No hay datos sobre el uso de este medicamento o de empagliflozina en mujeres embarazadas. Los estudios realizados en animales muestran que empagliflozina atraviesa la placenta durante la última fase de la gestación en un grado muy limitado, pero no indican efectos perjudiciales directos ni indirectos en lo que respecta al desarrollo embrionario temprano. No obstante, los estudios realizados en animales han mostrado efectos adversos en el desarrollo posnatal. Datos limitados sugieren que el uso de metformina en mujeres embarazadas no está asociado a un mayor riesgo de malformaciones congénitas. Los estudios realizados en animales con la combinación de empagliflozina y metformina o con metformina en monoterapia solo han mostrado toxicidad para la reproducción a dosis más altas de metformina (ver sección 5.3).
Cuando la paciente planea quedarse embarazada, y durante el embarazo, se recomienda que la diabetes no se trate con este medicamento, sino que se utilice insulina para mantener los niveles de glucosa en sangre lo más cercanos posible a los valores normales para disminuir el riesgo de malformaciones fetales asociadas a niveles anormales de glucosa en sangre.
Lactancia
Metformina se excreta en la leche materna. No se han observado efectos en niños/recién nacidos lactantes de mujeres tratadas con este medicamento. No se dispone de datos en humanos sobre la excreción de empagliflozina en la leche materna. Los datos disponibles en animales han mostrado que empagliflozina y metformina se excretan en la leche. No se puede excluir el riesgo para los recién nacidos o los lactantes.
Este medicamento no se debe utilizar durante la lactancia.
Fertilidad
No se han realizado estudios sobre el efecto de este medicamento o de empagliflozina en la fertilidad humana. Los estudios realizados en animales con empagliflozina y metformina no sugieren efectos perjudiciales directos o indirectos sobre la fertilidad (ver sección 5.3).
4.7 - Efectos sobre la capacidad de conducción de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
La influencia de Synjardy sobre la capacidad para conducir y utilizar máquinas es pequeña. Se debe advertir a los pacientes que tomen las debidas precauciones para evitar una hipoglucemia mientras conducen y utilizan máquinas, sobre todo cuando Synjardy se use en combinación con una sulfonilurea y/o con insulina.
4.8 - Reacciones Adversas de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Resumen del perfil de seguridad
Las reacciones adversas notificadas con más frecuencia en los ensayos clínicos fueron hipoglucemia en combinación con insulina y/o una sulfonilurea y síntomas gastrointestinales (náuseas, vómitos, diarrea, dolor abdominal y pérdida del apetito). No se identificaron reacciones adversas adicionales en los ensayos clínicos con empagliflozina como tratamiento adicional a metformina en comparación con las reacciones adversas de los componentes individuales.
Tabla de reacciones adversas
Las reacciones adversas se incluyen según la frecuencia absoluta. Las frecuencias se definen como muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1 000 a < 1/100), raras (≥ 1/10 000 a < 1/1 000), muy raras (< 1/10 000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 2. Tabla de reacciones adversas (MedDRA) procedentes de los ensayos controlados con placebo y de la experiencia poscomercialización
| Clasificación por órganos y sistemas | Muy frecuentes | Frecuentes | Poco frecuentes | Raras | Muy raras |
| Infecciones e infestaciones | Moniliasis vaginal, vulvovaginitis, balanitis y otras infecciones genitales1,2 Infecciones del tracto urinario (incluyendo pielonefritis y urosepsis)1,2 | Fascitis necrosante del perineo (gangrena de Fournier)a | |||
| Trastornos del metabolismo y de la nutrición | Hipoglucemia (cuando se usa con una sulfonilurea o con insulina)1 | Sed2 Disminución/ déficit de vitamina B123,a | Cetoacidosis diabéticaa | Acidosis láctica3 | |
| Trastornos del sistema nervioso | Alteraciones del gusto3 | ||||
| Trastornos vasculares | Hipovolemia1,2,d | ||||
| Trastornos gastrointestinales | Síntomas gastrointestinales3,4 | Estreñimiento | |||
| Trastornos hepatobiliares | Anomalías en las pruebas de la función hepática3 Hepatitis3 | ||||
| Trastornos de la piel y del tejido subcutáneo | Prurito (generalizado)2,3 Exantema | Urticaria Angioedema | Eritema3 | ||
| Trastornos renales y urinarios | Aumento de la micción1, 2 | Disuria2 | Nefritis tubulointersticial | ||
| Exploraciones complementarias | Aumento de los lípidos en suero2,b | Aumento de la creatinina en sangre/disminu ción de la tasa de filtración glomerular1 Aumento del hematocrito2,c |
1 Ver las subsecciones siguientes para obtener información adicional
2 Reacciones adversas identificadas con empagliflozina en monoterapia
3 Reacciones adversas identificadas con metformina en monoterapia
4 Los síntomas gastrointestinales como náuseas, vómitos, diarrea, dolor abdominal y pérdida del apetito se producen más frecuentemente al inicio del tratamiento y desaparecen de forma espontánea en la mayoría de los casos.
a Ver sección 4.4
b Los incrementos porcentuales medios respecto al valor basal para empagliflozina 10 mg y 25 mg frente a placebo, respectivamente, fueron del 5,0 % y 5,2 % frente al 3,7 % para el colesterol total; del 4,6 % y 2,7 % frente al -0,5 % para el colesterol HDL; del 9,1 % y 8,7 % frente al 7,8 % para el colesterol LDL; y del 5,4 % y 10,8 % frente al 12,1 % para los triglicéridos.
c Los cambios medios en el hematocrito respecto al valor basal fueron del 3,6 % y del 4,0 % para empagliflozina 10 mg y 25 mg, respectivamente, comparado con 0 % de placebo. En el ensayo EMPA-REG OUTCOME, los valores del hematocrito volvieron a los basales después de un período de seguimiento de 30 días tras finalizar el tratamiento.
d Los datos agrupados de los ensayos de empagliflozina en pacientes con insuficiencia cardíaca (en los que la mitad de los pacientes tenía diabetes mellitus de tipo 2) mostraron una frecuencia mayor de hipovolemia (“muy frecuente”: 11,4 % para empagliflozina frente al 9,7 % para el placebo).
Descripción de reacciones adversas seleccionadas
Hipoglucemia
La frecuencia de hipoglucemia dependió del tratamiento de base utilizado en los estudios correspondientes y fue similar en el caso de empagliflozina y del placebo como tratamiento adicional a metformina, como tratamiento adicional a linagliptina y metformina y para la combinación de empagliflozina con metformina en pacientes sin tratamiento previo en comparación con aquellos tratados con empagliflozina y metformina como componentes individuales, como asociado al tratamiento de referencia. Se observó un aumento de la frecuencia cuando empagliflozina se administró como tratamiento adicional a metformina y una sulfonilurea (empagliflozina 10 mg: 16,1 %, empagliflozina 25 mg: 11,5 % y placebo: 8,4 %), o como tratamiento adicional a metformina e insulina (empagliflozina 10 mg: 31,3 %, empagliflozina 25 mg: 36,2 % y placebo: 34,7 %).
Hipoglucemia grave (acontecimientos que requieran asistencia)
La frecuencia global de pacientes con acontecimientos hipoglucémicos graves fue baja (< 1 %) y similar en el caso de empagliflozina y del placebo como tratamiento adicional a metformina y para la combinación de empagliflozina con metformina en pacientes sin tratamiento previo en comparación con aquellos tratados con empagliflozina y metformina como componentes individuales, como asociado al tratamiento de referencia. Se produjeron acontecimientos hipoglucémicos graves en el 0,5 %, el 0 % y el 0,5 % de los pacientes tratados con empagliflozina 10 mg, empagliflozina 25 mg y placebo respectivamente cuando se utilizaron como tratamiento adicional a metformina e insulina.
Ningún paciente presentó acontecimientos hipoglucémicos graves en la combinación con metformina y una sulfonilurea y como tratamiento adicional a linagliptina y metformina.
Infecciones del tracto urinario
La frecuencia global de infecciones del tracto urinario notificadas como reacciones adversas fue mayor en los pacientes tratados con metformina que recibieron empagliflozina 10 mg (8,8 %) en comparación con los que recibieron empagliflozina 25 mg (6,6 %) o placebo (7,8 %). De manera similar al placebo, las infecciones del tracto urinario se notificaron con más frecuencia para empagliflozina en pacientes con antecedentes de infecciones crónicas o recurrentes del tracto urinario. La intensidad (leve, moderada, grave) de las infecciones del tracto urinario fue similar a la del placebo. Las infecciones del tracto urinario se notificaron con más frecuencia para empagliflozina 10 mg comparado con placebo en pacientes mujeres, pero esto no ocurrió para empagliflozina 25 mg. La frecuencia de las infecciones del tracto urinario fue baja en el caso de los pacientes varones y estuvo equilibrada en todos los grupos de tratamiento.
Moniliasis vaginal, vulvovaginitis, balanitis y otras infecciones genitales
La moniliasis vaginal, la vulvovaginitis, la balanitis y otras infecciones genitales se notificaron con más frecuencia en los pacientes tratados con metformina que recibieron empagliflozina 10 mg (4,0 %) y empagliflozina 25 mg (3,9 %) que en los que recibieron placebo (1,3 %) y se notificaron con más frecuencia pacientes mujeres tratadas con empagliflozina comparado con placebo. La diferencia en cuanto a frecuencia fue menos pronunciada en los pacientes varones. Las infecciones del tracto genital fueron de intensidad leve a moderada y ninguna tuvo una intensidad grave.
Aumento de la micción
Como cabe esperar del mecanismo de acción, el aumento de la micción (evaluado mediante una búsqueda de los términos predefinidos, incluyendo polaquiuria, poliuria y nocturia) se observó con mayor frecuencia en los pacientes tratados con metformina que recibieron empagliflozina 10 mg (3,0 %) y empagliflozina 25 mg (2,9 %) comparado con placebo (1,4 %) como tratamiento adicional a metformina. El aumento de la micción fue principalmente de intensidad leve a moderada. La frecuencia notificada de nocturia fue similar para placebo y para empagliflozina (< 1 %).
Hipovolemia
La frecuencia global de hipovolemia (incluyendo los términos predefinidos disminución (ambulatoria) de la presión arterial, disminución de la presión arterial sistólica, deshidratación, hipotensión, hipovolemia, hipotensión ortostática y síncope) en pacientes tratados con metformina que recibieron empagliflozina fue baja: 0,6 % en el caso de empagliflozina 10 mg, 0,3 % en el caso de empagliflozina 25 mg y 0,1 % en el caso del placebo. El efecto de empagliflozina en la eliminación de glucosa por la orina se asocia a la diuresis osmótica, que podría afectar al estado de hidratación de los pacientes de 75 años de edad o mayores. En pacientes ≥ 75 años de edad, los episodios de hipovolemia se han notificado en un único paciente tratado con empagliflozina como tratamiento adicional a metformina.
Aumento de la creatinina en sangre/Disminución de la tasa de filtración glomerular
La frecuencia general de pacientes con aumento de la creatinina en sangre y disminución de la tasa de filtración glomerular fue similar entre empaglifozina y placebo como tratamiento adicional a metformina (aumento de la creatinina en sangre: empagliflozina 10 mg 0,5 %, empagliflozina 25 mg 0,1 %, placebo 0,4 %; disminución de la tasa filtración glomerular: empagliflozina 10 mg 0,1 %, empagliflozina 25 mg 0 %, placebo 0,2 %).
Por lo general, los aumentos iniciales en la creatinina y las disminuciones iniciales en la tasa de filtración glomerular estimada en los pacientes tratados con empaglifozina fueron transitorios durante el tratamiento continuo o reversibles tras la suspensión del tratamiento con el medicamento.
De manera uniforme, en el ensayo EMPA-REG OUTCOME, los pacientes tratados con empagliflozina experimentaron un descenso inicial de la TFGe (media: 3 ml/min/1,73 m2). Posteriormente, la TFGe se mantuvo durante la continuación del tratamiento. La TFGe media recuperó el nivel basal tras la suspensión del tratamiento, lo cual indica que en estos cambios de la función renal podrían estar implicados cambios hemodinámicos agudos.
Población pediátrica
En el ensayo DINAMO se trató a 157 niños de 10 años de edad o mayores con diabetes tipo 2, de los cuales 52 pacientes recibieron empagliflozina, 52 recibieron linagliptina y 53 recibieron placebo (ver sección 5.1). Durante la fase controlada con placebo, la reacción adversa más frecuente fue la hipoglucemia (empagliflozina en dosis de 10 mg y 25 mg, datos agrupados: 23,1 %; placebo: 9,4 %). Ninguna de estas reacciones fue grave ni precisó asistencia.
En general, el perfil de seguridad en niños fue similar al perfil de seguridad en adultos con diabetes mellitus tipo 2.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es.
4.9 - Sobredosificación de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Síntomas
Empagliflozina
En ensayos clínicos controlados, dosis únicas de hasta 800 mg de empagliflozina (equivalente a 32 veces la dosis diaria máxima recomendada) en voluntarios sanos y dosis múltiples diarias de hasta 100 mg de empagliflozina (equivalente a 4 veces la dosis diaria máxima recomendada) en pacientes con diabetes tipo 2 no mostraron toxicidad. Empagliflozina aumentó la excreción de glucosa por la orina, lo que provocó un aumento en el volumen de orina. El aumento observado en el volumen de orina no fue dependiente de la dosis y no es clínicamente significativo. No hay experiencia con dosis superiores a 800 mg en humanos.
Metformina
No se ha observado hipoglucemia con dosis de metformina de hasta 85 g, aunque en estas circunstancias se ha producido acidosis láctica. Una sobredosis elevada de metformina o los riesgos concomitantes pueden producir acidosis láctica. La acidosis láctica es una emergencia médica que se debe tratar en un hospital (ver las secciones 4.4 y 4.5).
Tratamiento
En caso de sobredosis, se debe iniciar un tratamiento adecuado al estado clínico del paciente. El método más eficaz para eliminar el lactato y metformina es la hemodiálisis. No se ha estudiado la eliminación de empagliflozina mediante hemodiálisis.
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Grupo farmacoterapéutico: fármacos usados en diabetes, combinaciones de fármacos hipoglucemiantes orales, código ATC: A10BD20
Mecanismo de acción
Synjardy combina dos medicamentos hipoglucemiantes con mecanismos de acción complementarios para mejorar el control glucémico en pacientes con diabetes tipo 2: empagliflozina, un inhibidor del cotransportador de sodio-glucosa tipo 2 (SGLT2), e hidrocloruro de metformina, un miembro de la clase de las biguanidas.
Empagliflozina
Empagliflozina es un inhibidor competitivo reversible y selectivo altamente potente (IC50 de 1,3 nmol) del cotransportador de sodio-glucosa tipo 2 (SGLT2). Empagliflozina no inhibe otros transportadores de glucosa importantes para el transporte de glucosa a los tejidos periféricos y es 5 000 veces más selectiva para el SGLT2 que para el SGLT1, el transportador más importante responsable de la absorción de glucosa en el intestino. El SGLT2 se encuentra altamente expresado en el riñón, mientras que la expresión en otros tejidos es inexistente o muy baja. Es responsable, como transportador predominante, de la reabsorción de glucosa tras la filtración glomerular para devolverla a la circulación. En los pacientes con diabetes tipo 2 e hiperglucemia, se filtra y reabsorbe una mayor cantidad de glucosa.
Empagliflozina mejora el control glucémico en pacientes con diabetes tipo 2 al reducir la reabsorción renal de glucosa. La cantidad de glucosa eliminada por el riñón mediante este mecanismo glucurético depende de la concentración de glucosa en sangre y de la TFG. La inhibición del SLGT2 en pacientes con diabetes tipo 2 e hiperglucemia conduce a un exceso de excreción de glucosa por la orina.
Además, el inicio de la administración de empagliflozina aumenta la excreción de sodio, lo que da lugar a diuresis osmótica y a un volumen intravascular reducido.
En pacientes con diabetes tipo 2, la excreción de glucosa por la orina aumentó inmediatamente después de la primera dosis de empagliflozina y se mantuvo continua durante el intervalo de administración de 24 horas. El aumento en la eliminación de glucosa por la orina se mantuvo al final del periodo de tratamiento de 4 semanas, con un promedio de aproximadamente 78 g/día en el tratamiento con empagliflozina 25 mg. El aumento en la eliminación de glucosa por la orina dio lugar a una reducción inmediata de los niveles de glucosa plasmática en pacientes con diabetes tipo 2.
Empagliflozina mejora los niveles de glucosa en plasma, tanto en ayunas como posprandiales. El mecanismo de acción de empagliflozina es independiente de la función de las células beta y de la vía de la insulina, y esto contribuye a un bajo riesgo de hipoglucemia. Se observó una mejora de los marcadores indirectos de la función de las células beta, incluido el Modelo Homeostático β para la evaluación de la resistencia a la insulina (HOMA-β). Además, la excreción de glucosa por la orina desencadena una pérdida de calorías, que se asocia a una pérdida de grasa corporal y a una reducción de peso corporal. La glucosuria observada con empagliflozina se ve acompañada por una leve diuresis, que puede contribuir a la reducción sostenida y moderada de la presión arterial. La glucosuria, natriuresis y diuresis osmótica observadas con empagliflozina pueden contribuir a la mejora en los resultados cardiovasculares.
Metformina
Metformina es una biguanida con efectos antihiperglucemiantes que disminuye tanto la glucosa plasmática basal como la postprandial. No estimula la secreción de insulina y, por lo tanto, no provoca hipoglucemia.
Metformina puede actuar por 3 mecanismos:
- por disminución de la producción hepática de glucosa al inhibir la gluconeogénesis y la glucogenólisis,
- en el músculo, aumentando la sensibilidad a la insulina, mejorando la captación y utilización periféricas de glucosa,
- y retrasando la absorción intestinal de glucosa.
Metformina estimula la síntesis intracelular de glucógeno al actuar sobre la glucógeno-sintasa. Metformina aumenta la capacidad de transporte de todos los tipos de transportadores de glucosa de membrana (GLUTs) conocidos hasta ahora.
En los humanos, con independencia de su acción sobre la glucemia, metformina tiene efectos favorables sobre el metabolismo lipídico. Esto se ha demostrado a dosis terapéuticas en ensayos clínicos controlados a medio y largo plazo: metformina reduce los niveles de colesterol total, colesterol LDL y triglicéridos.
Eficacia clínica y seguridad
Tanto la mejora del control glucémico como la reducción de la morbilidad y mortalidad cardiovasculares son una parte integral del tratamiento de la diabetes tipo 2.
En 9 ensayos clínicos doble ciego, controlados con placebo y con tratamiento activo de al menos 24 semanas de duración, se han evaluado la eficacia glucémica y los resultados cardiovasculares en un total de 10 366 pacientes con diabetes tipo 2, de los cuales 2 950 recibieron empagliflozina 10 mg y 3 701 recibieron empagliflozina 25 mg como tratamiento adicional a metformina. De éstos, 266 o 264 pacientes se trataron con empagliflozina 10 mg o 25 mg como tratamiento adicional a metformina más insulina, respectivamente.
El tratamiento con empagliflozina en combinación con metformina, con o sin otros medicamentos antidiabéticos (pioglitazona, una sulfonilurea, inhibidores de la DPP-4 e insulina) proporcionó mejoras clínicamente significativas en la HbA1c, la glucosa plasmática en ayunas (GPA), el peso corporal y la presión arterial sistólica y diastólica. Con la administración de empagliflozina 25 mg, una mayor proporción de pacientes logró el objetivo de alcanzar una HbA1c inferior al 7 % y hubo menos pacientes que necesitaron un rescate glucémico en comparación con empagliflozina 10 mg y placebo. En pacientes de 75 años de edad o mayores, se observaron reducciones numéricamente más bajas en la HbA1c con el tratamiento con empagliflozina. Se asoció un nivel basal más alto de HbA1c con una mayor reducción de la HbA1c. Además, empagliflozina asociada al tratamiento de referencia redujo la mortalidad cardiovascular en pacientes con diabetes tipo 2 y enfermedad cardiovascular establecida.
Empagliflozina como tratamiento en combinación con metformina, sulfonilurea, pioglitazona
Empagliflozina como tratamiento adicional a metformina, metformina y una sulfonilurea o pioglitazona y metformina dio lugar a reducciones estadísticamente significativas (p < 0,0001) en la HbA1c y en el peso corporal comparado con placebo (Tabla 3). Además, también dio lugar a una reducción clínicamente significativa en la GPA y la presión arterial sistólica y diastólica comparado con placebo.
En la extensión doble ciego controlada con placebo de estos estudios, la reducción en la HbA1c, el peso corporal y la presión arterial se mantuvo hasta la semana 76.
Tabla 3. Resultados de eficacia de los ensayos controlados con placebo de 24 semanas
| Tratamiento adicional a metforminaa | ||||
| Placebo | Empagliflozina | |||
| 10 mg | 25 mg | |||
| N | 207 | 217 | 213 | |
| HbA1c (%) | ||||
| Valor basal (media) | 7,90 | 7,94 | 7,86 | |
| Cambio respecto al valor basal1 | -0,13 | -0,70 | -0,77 | |
| Diferencia con placebo1 (IC del 97,5 %) | -0,57* (-0,72, -0,42) | -0,64* (-0,79, -0,48) | ||
| N | 184 | 199 | 191 | |
| Pacientes (%) que alcanzan un valor de HbA1c < 7 % con un valor basal de la HbA1c ≥ 7 %2 | 12,5 | 37,7 | 38,7 | |
| N | 207 | 217 | 213 | |
| Peso corporal (kg) | ||||
| Valor basal (media) | 79,73 | 81,59 | 82,21 | |
| Cambio respecto al valor basal1 | -0,45 | -2,08 | -2,46 | |
| Diferencia con placebo1 (IC del 97,5 %) | -1,63* (-2,17, -1,08) | -2,01* (-2,56, -1,46) | ||
| N | 207 | 217 | 213 | |
| PAS (mm Hg)2 | ||||
| Valor basal (media) | 128,6 | 129,6 | 130,0 | |
| Cambio respecto al valor basal1 | -0,4 | -4,5 | -5,2 | |
| Diferencia con placebo1 (IC del 95 %) | -4,1* (-6,2, -2,1) | -4,8* (-6,9, -2,7) | ||
| Tratamiento adicional a metformina y una sulfonilureaa | ||||
| Placebo | Empagliflozina | |||
| 10 mg | 25 mg | |||
| N | 225 | 225 | 216 | |
| HbA1c (%) | ||||
| Valor basal (media) | 8,15 | 8,07 | 8,10 | |
| Cambio respecto al valor basal1 | -0,17 | -0,82 | -0,77 | |
| Diferencia con placebo1 (IC del 97,5 %) | -0,64* (-0,79, -0,49) | -0,59* (-0,74, -0,44) | ||
| N | 216 | 209 | 202 | |
| Pacientes (%) que alcanzan un valor de HbA1c < 7 % con un valor basal de la HbA1c ≥ 7 %2 | 9,3 | 26,3 | 32,2 | |
| N | 225 | 225 | 216 | |
| Peso corporal (kg) | ||||
| Valor basal (media) | 76,23 | 77,08 | 77,50 | |
| Cambio respecto al valor basal1 | -0,39 | -2,16 | -2,39 | |
| Diferencia con placebo1 (IC del 97,5 %) | -1,76* (-2,25, -1,28) | -1,99* (-2,48, -1,50) | ||
| N | 225 | 225 | 216 | |
| PAS (mm Hg)2 | ||||
| Valor basal (media) | 128,8 | 128,7 | 129,3 | |
| Cambio respecto al valor basal1 | -1,4 | -4,1 | -3,5 | |
| Diferencia con placebo1 (IC del 95 %) | -2,7 (-4,6, -0,8) | -2,1 (-4,0, -0,2) | ||
| Tratamiento adicional a pioglitazona + metforminab | ||||
| Placebo | Empagliflozina | |||
| 10 mg | 25 mg | |||
| N | 124 | 125 | 127 | |
| HbA1c (%) | ||||
| Valor basal (media) | 8,15 | 8,07 | 8,10 | |
| Cambio respecto al valor basal1 | -0,11 | -0,55 | -0,70 | |
| Diferencia con placebo1 (IC del 97,5 %) | -0,45* (-0,69, -0,21) | -0,60* (-0,83, -0,36) | ||
| N | 118 | 116 | 123 | |
| Pacientes (%) que alcanzan un valor de HbA1c < 7 % con un valor basal de la HbA1c ≥ 7 %2 | 8,5 | 22,4 | 28,5 | |
| N | 124 | 125 | 127 | |
| Peso corporal (kg) | ||||
| Valor basal (media) | 79,45 | 79,44 | 80,98 | |
| Cambio respecto al valor basal1 | 0,40 | -1,74 | -1,59 | |
| Diferencia con placebo1 (IC del 97,5 %) | -2,14* (-2,93, -1,35) | -2,00* (-2,78, -1,21) | ||
| N | 124 | 125 | 127 | |
| PAS (mm Hg)2, 3 | ||||
| Valor basal (media) | 125,5 | 126,3 | 126,3 | |
| Cambio respecto al valor basal1 | 0,8 | -3,5 | -3,3 | |
| Diferencia con placebo1 (IC del 95 %) | -4,2** (-6,94, -1,53) | -4,1** (-6,76, -1,37) | ||
a Grupo completo de análisis (FAS) utilizando la última observación considerada (LOCF) antes del tratamiento de rescate glucémico
b Análisis de subgrupos para pacientes con un tratamiento de base adicional con metformina (FAS, LOCF)
1 Media ajustada respecto al valor basal
2 No evaluado en cuanto a significación estadística como parte del procedimiento de prueba confirmatorio secuencial
3 LOCF, valores censurados estadísticamente después del rescate antihipertensivo
* valor p < 0,0001
** valor p < 0,01
Empagliflozina en combinación con metformina en pacientes sin tratamiento previo
Se realizó un estudio de diseño factorial de 24 semanas de duración para evaluar la eficacia y la seguridad de empagliflozina en pacientes sin tratamiento previo. El tratamiento con empagliflozina en combinación con metformina (5 mg y 500 mg; 5 mg y 1 000 mg; 12,5 mg y 500 mg; y 12,5 mg y 1 000 mg administrados dos veces al día) proporcionó mejoras estadísticamente significativas en la HbA1c (Tabla 4) y dio lugar a unas reducciones mayores en la GPA (en comparación con los componentes individuales) y en el peso corporal (en comparación con metformina).
Tabla 4. Resultados de eficacia en la semana 24 en los que se compara empagliflozina en combinación con metformina frente a los componentes individualesa
| Empagliflozina 10 mgb | Empagliflozina 25 mgb | Metforminac | |||||||
| +Met 1 000 mgc | +Met 2 000 mgc | Sin Met | +Met 1 000 mgc | +Met 2 000 mgc | Sin Met | 1 000 mg | 2 000 mg | ||
| N | 161 | 167 | 169 | 165 | 169 | 163 | 167 | 162 | |
| HbA1c (%) | |||||||||
| Valor basal (media) | 8,68 | 8,65 | 8,62 | 8,84 | 8,66 | 8,86 | 8,69 | 8,55 | |
| Cambio respecto al valor basal1 | -1,98 | -2,07 | -1,35 | -1,93 | -2,08 | -1,36 | -1,18 | -1,75 | |
| Comparación frente a empa (IC del 95 %)1 | -0,63* (-0,86, -0,40) | -0,72* (-0,96, -0,49) | -0,57* (-0,81, -0,34) | -0,72* (-0,95, -0,48) | |||||
| Comparación frente a met (IC del 95 %)1 | -0,79* (-1,03, -0,56) | -0,33* (-0,56, -0,09) | -0,75* (-0,98, -0,51) | -0,33* (-0,56, -0,10) | |||||
Met = metformina; empa = empagliflozina
1media ajustada respecto al valor basal
aLos análisis se realizaron en el grupo de análisis completo (GAC) utilizando un método de casos observados (CO)
bAdministrada en dos dosis iguales divididas al día cuando se administra con metformina
cAdministrada en dos dosis iguales divididas al día
*p ≤ 0,0062 para HbA1c
Empagliflozina en pacientes no controlados de forma adecuada con metformina y linagliptina
En pacientes no controlados de forma adecuada con metformina y linagliptina 5 mg, el tratamiento con ambas empagliflozina 10 mg y 25 mg dio lugar a reducciones estadísticamente significativas (p < 0,0001) en la HbA1c y en el peso corporal comparado con placebo (Tabla 5). Además, dio lugar a reducciones clínicamente significativas en la GPA y en la presión arterial sistólica y diastólica comparado con placebo.
Tabla 5. Resultados de eficacia de un ensayo controlado con placebo de 24 semanas en pacientes no controlados de forma adecuada con metformina y linagliptina 5 mg
| Tratamiento adicional a metformina y linagliptina 5 mg | ||||
| Placebo5 | Empagliflozina6 | |||
| 10 mg | 25 mg | |||
| N | 106 | 109 | 110 | |
| HbA1c (%)3 | ||||
| Valor basal (media) | 7,96 | 7,97 | 7,97 | |
| Cambio respecto al valor basal1 | 0,14 | -0,65 | -0,56 | |
| Diferencia con placebo (IC del 95 %) | -0,79* (-1,02, -0,55) | -0,70* (-0,93, -0,46) | ||
| N | 100 | 100 | 107 | |
| Pacientes (%) que alcanzan un valor de HbA1c < 7 % con un valor basal de HbA1c ≥ 7 %2 | 17,0 | 37,0 | 32,7 | |
| N | 106 | 109 | 110 | |
| Peso corporal (kg)3 | ||||
| Valor basal (media) | 82,3 | 88,4 | 84,4 | |
| Cambio respecto al valor basal1 | -0,3 | -3,1 | -2,5 | |
| Diferencia con placebo (IC del 95 %) | -2,8* (-3,5, -2,1) | -2,2* (-2,9, -1,5) | ||
| N | 106 | 109 | 110 | |
| PAS (mm Hg)4 | ||||
| Valor basal (media) | 130,1 | 130,4 | 131,0 | |
| Cambio respecto al valor basal1 | -1,7 | -3,0 | -4,3 | |
| Diferencia con placebo (IC del 95 %) | -1,3 (-4,2, 1,7) | -2,6 (-5,5, 0,4) | ||
1 Media ajustada respecto al valor basal
2 No evaluado en cuanto a significación estadística; no forma parte del procedimiento de prueba secuencial para las variables secundarias
3 El modelo mixto de medidas repetidas (MMRM) en el GAC (CO) incluyó HbA1c basal, TFGe basal (modificación de la dieta en el estudio de la enfermedad renal, MDRD), región geográfica, visita, tratamiento y tratamiento por interacción de la visita. Para el peso, se incluyó el peso basal.
4 El modelo mmRM incluyó la PAS basal y la HbA1c basal como covariable(s) lineal(es), y la TFGe basal, región geográfica, tratamiento, visita y visita por interacción con el tratamiento como efectos fijos.
5 Los pacientes aleatorizados al grupo de placebo recibieron placebo más linagliptina 5 mg con tratamiento de base de metformina
6 Los pacientes aleatorizados a los grupos de empagliflozina 10 mg o 25 mg recibieron empagliflozina 10 mg o 25 mg y linagliptina 5 mg con tratamiento de base de metformina
* Valor de p < 0,0001
En un subgrupo pre-especificado de pacientes con HbA1c basal mayor o igual al 8,5 % la reducción con respecto al valor basal en la HbA1c fue del -1.3 % con empagliflozina 10 mg o 25 mg a las 24 semanas (p < 0,0001) comparado con placebo.
Datos de empagliflozina de 24 meses, como tratamiento adicional a metformina en comparación con glimepirida
En un ensayo que comparó la eficacia y la seguridad de empagliflozina 25 mg frente a glimepirida (hasta 4 mg al día) en pacientes con un control glucémico inadecuado con metformina sola, el tratamiento diario con empagliflozina dio lugar a una mayor reducción de la HbA1c (Tabla 6) y a una reducción clínicamente significativa de la GPA en comparación con glimepirida. La administración diaria de empagliflozina provocó una reducción estadísticamente significativa del peso corporal y de la presión arterial sistólica y diastólica, y una proporción inferior de pacientes estadísticamente significativa con episodios hipoglucémicos en comparación con glimepirida (2,5 % para empagliflozina, 24,2 % para glimepirida, p < 0,0001).
Tabla 6. Resultados de eficacia en la semana 104 de un ensayo controlado con activo que comparó empagliflozina con glimepirida como tratamiento adicional a metforminaa
| Empagliflozina 25 mg | Glimepiridab | ||
| N | 765 | 780 | |
| HbA1c (%) | |||
| Valor basal (media) | 7,92 | 7,92 | |
| Cambio respecto al valor basal1 | -0,66 | -0,55 | |
| Diferencia respecto a la glimepirida1 (IC del 97,5 %) | -0,11* (-0,20, -0,01) | ||
| N | 690 | 715 | |
| Pacientes (%) que alcanzan un valor de HbA1c < 7 % con un valor basal de la HbA1c ≥ 7 %2 | 33,6 | 30,9 | |
| N | 765 | 780 | |
| Peso corporal (kg) | |||
| Valor basal (media) | 82,52 | 83,03 | |
| Cambio respecto al valor basal1 | -3,12 | 1,34 | |
| Diferencia respecto a la glimepirida1 (IC del 97,5 %) | -4,46** (-4,87, -4,05) | ||
| N | 765 | 780 | |
| PAS (mm Hg)3 | |||
| Valor basal (media) | 133,4 | 133,5 | |
| Cambio respecto al valor basal1 | -3,1 | 2,5 | |
| Diferencia respecto a la glimepirida1 (IC del 97,5 %) | -5,6** (-7,0,-4,2) | ||
a Grupo completo de análisis (FAS) utilizando la última observación considerada (LOCF) antes del tratamiento de rescate glucémico
b Hasta 4 mg de glimepirida
1 Media ajustada respecto al valor basal
2 No evaluado en cuanto a significación estadística como parte del procedimiento de prueba confirmatorio secuencial
3 LOCF, valores censurados estadísticamente después del rescate antihipertensivo
* valor p < 0,0001 para no inferioridad, y valor p = 0,0153 para superioridad
** valor p < 0,0001
Tratamiento adicional a insulina
Empagliflozina como tratamiento adicional a dosis diarias múltiples de insulina
Se evaluó la eficacia y la seguridad de empagliflozina como tratamiento adicional a dosis diarias múltiples de insulina con metformina concomitante en un ensayo doble ciego controlado con placebo de 52 semanas de duración. Durante las 18 primeras semanas y las 12 últimas semanas, la dosis de insulina se mantuvo estable, pero se ajustó entre las semanas 19 y 40 para alcanzar niveles de glucosa preprandial < 100 mg/dl [5,5 mmol/l] y niveles de glucosa posprandial < 140 mg/dl [7,8 mmol/l].
En la semana 18, empagliflozina presentó una mejora estadísticamente significativa de la HbA1c comparado con placebo (Tabla 7).
En la semana 52, el tratamiento con empagliflozina provocó una reducción estadísticamente significativa de la HbA1c y un ahorro de insulina comparado con placebo, así como una reducción en el peso corporal.
Tabla 7. Resultados de eficacia en las semanas 18 y 52 de un ensayo controlado con placebo de empagliflozina como tratamiento adicional a dosis múltiples diarias de insulina con tratamiento concomitante con metformina
| Placebo | Empagliflozina | |||
| 10 mg | 25 mg | |||
| N | 135 | 128 | 137 | |
| HbA1c (%) en la semana 18a | ||||
| Valor basal (media) | 8,29 | 8,42 | 8,29 | |
| Cambio respecto al valor basal1 | -0,58 | -0,99 | -1,03 | |
| Diferencia con placebo1 (IC del 97,5 %) | -0,41* (-0,61, -0,21) | -0,45* (-0,65, -0,25) | ||
| N | 86 | 84 | 87 | |
| HbA1c (%) en la semana 52b | ||||
| Valor basal (media) | 8,26 | 8,43 | 8,38 | |
| Cambio respecto al valor basal1 | -0,86 | -1,23 | -1,31 | |
| Diferencia con placebo1 (IC del 97,5 %) | -0,37** (-0,67, -0,08) | -0,45* (-0,74, -0,16) | ||
| N | 84 | 84 | 87 | |
| Pacientes (%) que alcanzan un valor de la HbA1c < 7 % con un valor basal de la HbA1c ≥ 7 % en la semana 52b,2 | 27,4 | 41,7 | 48,3 | |
| N | 86 | 83 | 86 | |
| Valor basal (media) | 91,01 | 91,77 | 90,22 | |
| Cambio respecto al valor basal1 | 12,84 | 0,22 | -2,25 | |
| Diferencia con placebo1 (IC del 97,5 %) | -12,61** (-21,43, -3,80) | -15,09** (-23,79, -6,40) | ||
| N | 86 | 84 | 87 | |
| Valor basal (media) | 97,78 | 98,86 | 94,93 | |
| Cambio respecto al valor basal1 | 0,42 | -2,47 | -1,94 | |
| Diferencia con placebo1 (IC del 97,5 %) | -2,89* (-4,29, -1,49) | -2,37* (-3,75, -0,98) | ||
a Análisis de subgrupos para pacientes con un tratamiento de base adicional con metformina (FAS, LOCF)
b Análisis de subgrupos para pacientes con un tratamiento de base adicional con metformina (pacientes que completaron el estudio de la población por protocolo (PPS), LOCF)
1 Media ajustada respecto al valor basal
2 No evaluado en cuanto a significación estadística como parte del procedimiento de prueba confirmatorio secuencial
3 Semana 19-40: pauta de tratamiento hasta alcanzar el objetivo con ajuste de la dosis de insulina para conseguir niveles objetivo de glucosa predefinidos (preprandial < 100 mg/dl (5,5 mmol/l), posprandial < 140 mg/dl (7,8 mmol/l)
* valor p ≤ 0,0005
** valor p < 0,005
Empagliflozina como tratamiento adicional a insulina basal
Se evaluó la eficacia y la seguridad de empagliflozina como tratamiento adicional a insulina basal con tratamiento concomitante con metformina en un ensayo doble ciego controlado con placebo de 78 semanas de duración. Durante las 18 primeras semanas, la dosis de insulina se mantuvo estable, pero se ajustó para lograr una GPA < 110 mg/dl en las 60 semanas siguientes.
En la semana 18, empagliflozina presentó una mejora estadísticamente significativa en la HbA1c. Una proporción mayor de pacientes tratados con empagliflozina y con un valor basal de la HbA1c ≥ 7,0 % lograron el valor objetivo de la HbA1c de < 7 % comparado con placebo (Tabla 8).
En la semana 78, se mantuvieron la disminución de la HbA1c y el ahorro de insulina debido a empagliflozina. Además, empagliflozina dio lugar a una reducción de la GPA, el peso corporal y la presión arterial.
Tabla 8. Resultados de eficacia en las semanas 18 y 78 de un ensayo controlado con placebo de empagliflozina como tratamiento adicional a insulina basal con metforminaa
| Placebo | Empagliflozina 10 mg | Empagliflozina 25 mg | ||
| N | 96 | 107 | 99 | |
| HbA1c (%) en la semana 18 | ||||
| Valor basal (media) | 8,02 | 8,21 | 8,35 | |
| Cambio respecto al valor basal1 | -0,09 | -0,62 | -0,72 | |
| Diferencia con placebo1 (IC del 97,5 %) | -0,54* (-0,77, -0,30) | -0,63* (-0,88, -0,39) | ||
| N | 89 | 105 | 94 | |
| HbA1c (%) en la semana 78 | ||||
| Valor basal (media) | 8,03 | 8,24 | 8,29 | |
| Cambio respecto al valor basal1 | -0,08 | -0,42 | -0,71 | |
| Diferencia con placebo1 (IC del 97,5 %) | -0,34** (-0,64, -0,05) | -0,63* (-0,93, -0,33) | ||
| N | 89 | 105 | 94 | |
| Dosis de insulina basal (UI/día) en la semana 78 | ||||
| Valor basal (media) | 49,61 | 47,25 | 49,37 | |
| Cambio respecto al valor basal1 | 4,14 | -2,07 | -0,28 | |
| Diferencia con placebo1 (IC del 97,5 %) | -6,21** (-11,81, -0,61) | -4,42 (-10,18, 1,34) | ||
a Análisis de subgrupos del grupo completo de análisis (FAS) para pacientes con un tratamiento de base adicional con metformina: pacientes que completaron el estudio utilizando la última observación considerada (LOCF) antes del tratamiento de rescate glucémico
1 Media ajustada respecto al valor basal
* valor p < 0,0001
** valor p ≤ 0,025
Empagliflozina y linagliptina como tratamiento adicional a metformina
En un ensayo doble ciego realizado en pacientes con un control glucémico inadecuado, el tratamiento de 24 semanas con dosis de ambas empagliflozina más linagliptina como tratamiento adicional a metformina provocó reducciones estadísticamente significativas (p < 0,0001) en la HbA1c (cambio respecto al valor basal de -1,08 % en el caso de empagliflozina 10 mg más linagliptina 5 mg, -1,19 % en el caso de empagliflozina 25 mg más linagliptina 5 mg, -0,70 % en el caso de linagliptina 5 mg). En comparación con linagliptina 5 mg, las dos dosis de empagliflozina más linagliptina 5 mg provocaron reducciones estadísticamente significativas de la GPA y la presión arterial. Las dos dosis provocaron reducciones estadísticamente significativas similares del peso corporal, expresado en kg y como porcentaje de cambio. Una proporción mayor de pacientes con un valor basal de la HbA1c ≥ 7,0 % y tratados con empagliflozina más linagliptina lograron el valor objetivo de la HbA1c de < 7 % en comparación con linagliptina 5 mg. Las reducciones clínicamente significativas de la HbA1c se mantuvieron durante 52 semanas.
Empagliflozina dos veces al día frente a una vez al día como tratamiento adicional a metformina
Se evaluó la eficacia y la seguridad de empagliflozina dos veces al día frente a una vez al día (dosis diaria de 10 mg y 25 mg) como tratamiento adicional en pacientes con un control glucémico suficiente que estaban recibiendo metformina en monoterapia en un ensayo doble ciego controlado con placebo de 16 semanas de duración. Todos los tratamientos con empagliflozina provocaron reducciones estadísticamente significativas de la HbA1c con respecto al valor basal (media total del 7,8 %) después de 16 semanas de tratamiento comparado con placebo. Las pautas posológicas de empagliflozina dos veces al día con un tratamiento de base con metformina provocaron reducciones similares en la HbA1c a las pautas posológicas de una vez al día, con una diferencia del tratamiento en las reducciones de la HbA1c respecto al valor basal hasta la semana 16 del -0,02 % (IC del 95 %, -0,16, 0,13) en el caso de empagliflozina 5 mg dos veces al día frente a 10 mg una vez al día, y del -0,11 % (IC del 95 %, -0,26, 0,03) en el caso de empagliflozina 12,5 mg dos veces al día frente a 25 mg una vez al día.
Resultados cardiovasculares
El ensayo doble ciego controlado con placebo EMPA-REG OUTCOME comparó dosis agrupadas de empagliflozina 10 mg y 25 mg con placebo asociadas al tratamiento de referencia en pacientes con diabetes tipo 2 y enfermedad cardiovascular establecida. Se trató a un total de 7 020 pacientes (empagliflozina 10 mg: 2 345, empagliflozina 25 mg: 2 342, placebo: 2 333) y se les realizó seguimiento durante una media de 3,1 años. La edad media fue de 63 años, la media de la HbA1c fue del 8,1 %, y el 71,5 % eran varones. Al inicio, el 74 % de los pacientes se estaban tratando con metformina, el 48 % con insulina y el 43 % con una sulfonilurea. Alrededor de la mitad de los pacientes (52,2 %) tenían una TFGe de 60-90 ml/min/1,73 m2, el 17,8 % de 45-60 ml/min/1,73 m2 y el 7,7 % de 30-45 ml/min/1,73 m2.
En la semana 12 se observó una mejora de la media ajustada (DE) en la HbA1c, cuando se comparó con el valor basal, del 0,11 % (0,02) en el grupo de placebo, y del 0,65 % (0,02) y 0,71 % (0,02) en los grupos de empagliflozina 10 y 25 mg, Tras las primeras 12 semanas, el control glucémico se optimizó de manera independiente del tratamiento en investigación. Por tanto, el efecto se atenuó en la semana 94, con una mejora de la media ajustada (DE) en la HbA1c del 0,08 % (0,02) en el grupo de placebo, y del 0,50 % (0,02) y 0,55 % (0,02) en los grupos de empagliflozina 10 y 25 mg.
Empagliflozina fue superior en prevenir la variable primaria combinada de muerte cardiovascular, infarto de miocardio no mortal o accidente cerebrovascular no mortal comparado con placebo. El efecto del tratamiento se observó mediante una reducción significativa en la muerte cardiovascular sin cambio significativo en el infarto de miocardio no mortal o en el accidente cerebrovascular no mortal.
La reducción de la muerte cardiovascular fue comparable para empagliflozina 10 mg y 25 mg (Figura 1) y confirmada mediante una mejora de la supervivencia global (Tabla 9). El efecto de empagliflozina sobre la variable primaria compuesta de muerte cardiovascular, infarto de miocardio no mortal o accidente cerebrovascular no mortal fue en gran medida independiente del control glucémico o de la función renal (TFGe) y, en general, similar en las distintas categorías de TFGe hasta una TFGe de 30 ml/min/1,73 m2 en el estudio EMPA-REG OUTCOME.
La eficacia en la prevención de la mortalidad cardiovascular no se ha establecido de manera concluyente en pacientes tratados con empagliflozina de forma concomitante con inhibidores de la DPP-4 o en pacientes de raza negra debido a que la representación de estos grupos en el estudio EMPA-REG OUTCOME fue limitada.
Tabla 9. Efecto del tratamiento para la variable primaria compuesta, sus componentes y mortalidada
| Placebo | Empagliflozinab | |
| N | 2 333 | 4 687 |
| Tiempo hasta el primer acontecimiento de muerte CV, IM no mortal o accidente cerebrovascular no mortal) N (%) | 282 (12,1) | 490 (10,5) |
| Cociente de riesgo instantáneo frente a placebo (IC del 95,02 %)* | 0,86 (0,74, 0,99) | |
| Valor p para la superioridad | 0,0382 | |
| Muerte CV N (%) | 137 (5,9) | 172 (3,7) |
| Cociente de riesgo instantáneo frente a placebo (IC del 95 %) | 0,62 (0,49, 0,77) | |
| Valor p | <0,0001 | |
| IM no mortal N (%) | 121 (5,2) | 213 (4,5) |
| Cociente de riesgo instantáneo frente a placebo (IC del 95 %) | 0,87 (0,70, 1,09) | |
| Valor p | 0,2189 | |
| Accidente cerebrovascular no mortal N (%) | 60 (2,6) | 150 (3,2) |
| Cociente de riesgo instantáneo frente a placebo (IC del 95 %) | 1,24 (0,92, 1,67) | |
| Valor p | 0,1638 | |
| Mortalidad por todas las causas N (%) | 194 (8,3) | 269 (5,7) |
| Cociente de riesgo instantáneo frente a placebo (IC del 95 %) | 0,68 (0,57, 0,82) | |
| Valor p | <0,0001 | |
| Mortalidad no CV N (%) | 57 (2,4) | 97 (2,1) |
| Cociente de riesgo instantáneo frente a placebo (IC del 95 %) | 0,84 (0,60, 1,16) |
CV = cardiovascular, IM = infarto de miocardio
a Conjunto tratado (CT), es decir, pacientes que han recibido al menos una dosis del medicamento del estudio
b Dosis agrupadas de empagliflozina 10 mg y 25 mg
* Puesto que los datos del ensayo se incluyeron en un análisis intermedio, se aplica un intervalo de confianza bilateral del 95,02 % que corresponde a un valor p menor de 0,0498 para la significación.
Figura 1. Tiempo hasta la aparición de muerte cardiovascular en el ensayo EMPA-REG OUTCOME
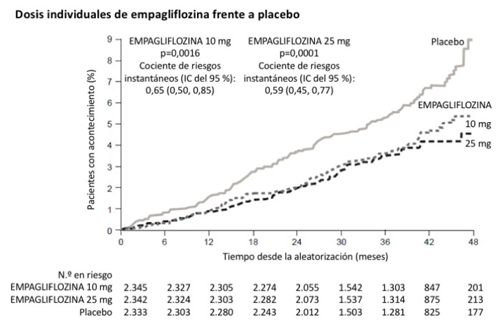
Insuficiencia cardíaca que requiere hospitalización
En el ensayo EMPA-REG OUTCOME, empagliflozina redujo el riesgo de hospitalización por insuficiencia cardíaca en comparación con el placebo (empagliflozina 2,7 %; placebo 4,1 %. Razón de riesgos instantáneos [RRI] 0,65, IC del 95 % 0,50, 0,85).
Nefropatía
En el ensayo EMPA-REG OUTCOME, en relación al tiempo hasta el primer acontecimiento de nefropatía, la RRI fue de 0,61 (IC del 95 % 0,53, 0,70) para empagliflozina (12,7 %) en comparación con el placebo (18,8 %).
Además, empagliflozina mostró una incidencia mayor (RRI 1,82, IC del 95 % 1,40, 2,37) de normoalbuminuria o microalbuminuria sostenida (49,7 %) en los pacientes con macroalbuminuria basal en comparación con el placebo (28,8 %).
Glucosa posprandial a las 2 horas
El tratamiento con empagliflozina como tratamiento adicional a metformina o metformina más una sulfonilurea provocó una mejora clínicamente significativa de la glucosa posprandial a las 2 horas (prueba de tolerancia a la glucosa) a las 24 semanas (tratamiento adicional a metformina: placebo +5,9 mg/dl, empagliflozina 10 mg: -46,0 mg/dl, empagliflozina 25 mg: -44,6 mg/dl, tratamiento adicional a metformina más una sulfonilurea, placebo: -2,3 mg/dl, empagliflozina 10 mg: -35,7 mg/dl, empagliflozina 25 mg: -36,6 mg/dl).
Pacientes con un valor basal de la HbA1c ≥ 9 %
En un análisis preespecificado de individuos con un valor basal de la HbA1c ≥ 9,0 %, el tratamiento con empagliflozina 10 mg o 25 mg como tratamiento adicional a metformina provocó reducciones estadísticamente significativas de la HbA1c en la semana 24 (cambio medio ajustado respecto al valor basal de -1,49 % en el caso de empagliflozina 25 mg, -1,40 % en el caso de empagliflozina 10 mg y -0,44 % en el caso del placebo).
Peso corporal
En un análisis conjunto preespecificado de 4 ensayos controlados con placebo, el tratamiento con empagliflozina (donde el 68 % de todos los pacientes estaban recibiendo tratamiento de base con metformina) provocó una reducción del peso corporal comparado con placebo en la semana 24 (-2,04 kg en el caso de empagliflozina 10 mg, -2,26 kg en el caso de empagliflozina 25 mg y -0,24 kg en el caso del placebo), que se mantuvo hasta la semana 52 (-1,96 kg en el caso de empagliflozina 10 mg, -2,25 kg en el caso de empagliflozina 25 mg y -0,16 kg en el caso de placebo).
Presión arterial
La eficacia y la seguridad de empagliflozina se evaluaron en un ensayo doble ciego controlado con placebo de 12 semanas de duración, en pacientes con diabetes tipo 2 y presión arterial alta que seguían diferentes tratamientos antidiabéticos y hasta 2 tratamientos antihipertensivos. El tratamiento con empagliflozina una vez al día provocó una mejora estadísticamente significativa en la HbA1c, y en la presión arterial sistólica y diastólica media de 24 horas, determinada mediante controles ambulatorios de la presión arterial (Tabla 10). El tratamiento con empagliflozina provocó reducciones en la PAS y PAD en posición sentada.
Tabla 10. Resultados de eficacia en la semana 12 de un ensayo controlado con placebo de empagliflozina en pacientes con diabetes tipo 2 y presión arterial no controladaa
| Placebo | Empagliflozina | |||
| 10 mg | 25 mg | |||
| N | 271 | 276 | 276 | |
| HbA1c (%) en la semana 121 | ||||
| Valor basal (media) | 7,90 | 7,87 | 7,92 | |
| Cambio respecto al valor basal2 | 0,03 | -0,59 | -0,62 | |
| Diferencia con placebo1 (IC del 95 %)2 | -0,62* (-0,72, -0,52) | -0,65* (-0,75, -0,55) | ||
| PAS a las 24 horas en la semana 123 | ||||
| Valor basal (media) | 131,72 | 131,34 | 131,18 | |
| Cambio respecto al valor basal4 | 0,48 | -2,95 | -3,68 | |
| Diferencia con placebo4 (IC del 95 %) | -3,44* (-4,78, -2,09) | -4,16* (-5,50, -2,83) | ||
| PAD a las 24 horas en la semana 123 | ||||
| Valor basal (media) | 75,16 | 75,13 | 74,64 | |
| Cambio respecto al valor basal5 | 0,32 | -1,04 | -1,40 | |
| Diferencia con placebo5 (IC del 95 %) | -1,36** (-2,15, -0,56) | -1,72* (-2,51, -0,93) | ||
a Grupo completo de análisis (FAS)
1 LOCF, valores censurados estadísticamente después del tratamiento de rescate antidiabético
2 Media ajustada respecto a la HbA1c basal, la TFGe basal, la región geográfica y el número de medicamentos antihipertensivos
3 LOCF, valores censurados estadísticamente después del tratamiento de rescate antidiabético o de cambiar el tratamiento de rescate antihipertensivo
4 Media ajustada respecto a la PAS basal la HbA1c basal, la TFGe basal, la región geográfica y el número de medicamentos antihipertensivos
5 Media ajustada respecto a la PAD basal, la HbA1c basal, la TFGe basal, la región geográfica y el número de medicamentos antihipertensivos
* valor p < 0,0001
** valor p < 0,001
En un análisis conjunto preespecificado de 4 ensayos controlados con placebo, el tratamiento con empagliflozina (donde el 68 % de todos los pacientes estaba recibiendo tratamiento de base con metformina) provocó una reducción de la presión arterial sistólica (empagliflozina 10 mg: -3,9 mm Hg; empagliflozina 25 mg: -4,3 mm Hg) comparado con placebo (- 0,5 mm Hg) y de la presión arterial diastólica (empagliflozina 10 mg: -1,8 mm Hg; empagliflozina 25 mg: -2,0 mm Hg) comparado con placebo (-0,5 mm Hg) en la semana 24 y esta reducción se mantuvo hasta la semana 52.
Metformina
El estudio prospectivo aleatorizado (UKPDS) ha establecido el efecto beneficioso a largo plazo del control intensivo de la glucemia en la diabetes tipo 2. El análisis de los resultados obtenidos en pacientes con sobrepeso tratados con metformina tras el fracaso de la dieta sola demostró:
- una reducción significativa del riesgo absoluto de complicaciones relacionadas con la diabetes en el grupo de metformina (29,8 acontecimientos/1 000 paciente-años) frente a dieta sola
(43,3 acontecimientos/1 000 paciente-años), p = 0,0023, y frente a los grupos de tratamiento combinado con sulfonilurea e insulina en monoterapia
(40,1 acontecimientos/1 000 paciente-años), p = 0,0034, - una reducción significativa del riesgo absoluto de cualquier mortalidad relacionada con la diabetes: metformina: 7,5 acontecimientos/1000 paciente-años, dieta sola:
12,7 acontecimientos/1 000 paciente-años, p = 0,017, - una reducción significativa del riesgo absoluto de mortalidad global: metformina: 13,5 acontecimientos/1 000 paciente-años frente a dieta sola:
20,6 acontecimientos/1 000 paciente-años, (p = 0,011) y frente a los grupos de tratamiento combinado con sulfonilurea e insulina en monoterapia:
18,9 acontecimientos/1 000 paciente-años (p = 0,021), - una reducción significativa del riesgo absoluto de infarto de miocardio: metformina: 11 acontecimientos/1 000 paciente-años, dieta sola: 18 acontecimientos/1 000 paciente-años, (p = 0,01).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Synjardy en todos los grupos de la población pediátrica desde el nacimiento hasta menos de 10 años de edad en diabetes tipo 2 (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
Se ha evaluado la eficacia clínica y la seguridad de empagliflozina (10 mg con un posible aumento de la dosis a 25 mg) y linagliptina (5 mg) una vez al día en niños y adolescentes de entre 10 y 17 años de edad con diabetes mellitus tipo 2 en un estudio controlado con placebo (DINAMO) durante 26 semanas, con un periodo de extensión de seguridad de hasta 52 semanas. Entre los tratamientos de base como complemento de la dieta y el ejercicio se incluyeron metformina (51 %), una combinación de metformina e insulina (40,1 %), insulina (3,2 %) o ninguno (5,7 %).
El cambio medio ajustado de la HbA1c en la semana 26 entre empagliflozina (N = 52) y placebo (N = 53) de –0,84 % fue clínicamente importante y estadísticamente significativo (IC del 95 % –1,50, –0,19; p = 0,0116). Además, el tratamiento con empagliflozina en comparación con placebo produjo un cambio medio ajustado clínicamente importante de la GPA de –35,2 mg/dl (IC del 95 % –58,6, – 11,7) (–1,95 mmol/l [–3,25, –0,65]). En el subgrupo de metformina (empagliflozina, N = 48; placebo, N = 47), los cambios fueron de –0,76 % (IC del 95 % –1,45 %, –0,08 %) para la HbA1c y de – 38,28 mg/dl (IC del 95 % –60,47, –16,10) para la GPA.
5.2 - Propiedades farmacocinéticas de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Synjardy
Los resultados de los estudios de bioequivalencia realizados en sujetos sanos demostraron que los comprimidos combinados de Synjardy (empagliflozina/hidrocloruro de metformina) 5 mg/850 mg, 5 mg/1 000 mg, 12,5 mg/850 mg y 12,5 mg/1 000 mg son bioequivalentes a la administración conjunta de las dosis correspondientes de empagliflozina y metformina en comprimidos distintos.
La administración de empagliflozina/metformina 12,5 mg/1 000 mg en situación posprandial dio lugar a una disminución del 9 % en el AUC y a una disminución del 28 % en la Cmax de empagliflozina, en comparación con la administración en ayunas. En el caso de metformina, el AUC disminuyó en un 12 % y la Cmax disminuyó en un 26 % en comparación con la administración en ayunas. El efecto observado de los alimentos sobre la farmacocinética de empagliflozina y metformina no se considera clínicamente relevante. No obstante, puesto que se recomienda administrar metformina con las comidas, también se propone administrar Synjardy con alimentos.
A continuación se reflejan las propiedades farmacocinéticas de cada uno de los principios activos de Synjardy.
Empagliflozina
Absorción
La farmacocinética de empagliflozina se ha caracterizado extensivamente en voluntarios sanos y en pacientes con diabetes tipo 2. Tras la administración oral, empagliflozina se absorbió rápidamente, alcanzándose concentraciones plasmáticas máximas a una mediana de tmax de 1,5 horas después de la dosis. Después, las concentraciones plasmáticas disminuyeron de forma bifásica con una fase de distribución rápida y una fase terminal relativamente lenta. La AUC plasmática media en estado estacionario y la Cmax fueron de 1 870 nmol.h/l y 259 nmol/l con empagliflozina 10 mg y de 4 740 nmol.h/l y 687 nmol/l con empagliflozina 25 mg una vez al día. La exposición sistémica de empagliflozina aumentó de forma proporcional a la dosis. Los parámetros farmacocinéticos de dosis única y de estado estacionario de empagliflozina fueron similares, lo que sugiere una farmacocinética lineal respecto al tiempo. No hubo diferencias clínicamente relevantes en la farmacocinética de empagliflozina entre los voluntarios sanos y los pacientes con diabetes tipo 2.
Se comparó la farmacocinética de 5 mg de empagliflozina dos veces al día y 10 mg de empagliflozina una vez al día en sujetos sanos. La exposición global (AUCss) de empagliflozina durante un período de 24 horas con la administración de empagliflozina 5 mg dos veces al día fue similar a la administración de empagliflozina 10 mg una vez al día. Tal como se esperaba, la administración de empagliflozina 5 mg dos veces al día dio lugar a una Cmax más baja y a unas concentraciones plasmáticas mínimas de empagliflozina más altas (Cmin) en comparación con empagliflozina 10 mg una vez al día.
La administración de empagliflozina 25 mg después de la ingesta de una comida rica en grasas y alta en calorías dio lugar a una exposición ligeramente inferior; la AUC disminuyó en aproximadamente el 16 % y la Cmax disminuyó en aproximadamente un 37 % en comparación con las condiciones de ayunas. El efecto observado de los alimentos sobre la farmacocinética de empagliflozina no se consideró clínicamente relevante, por lo que empagliflozina se puede administrar con o sin alimentos. Se obtuvieron resultados similares cuando los comprimidos combinados de Synjardy (empagliflozina/metformina) se administraron con una comida rica en grasas y alta en calorías.
Distribución
De acuerdo al análisis farmacocinético poblacional, se calculó que el volumen de distribución aparente en estado estacionario era de 73,8 litros. Después de la administración de una solución oral de [14C]-empagliflozina a voluntarios sanos, la distribución de los glóbulos rojos fue de aproximadamente un 37 % y la unión a proteínas plasmáticas, del 86 %.
Biotransformación
No se detectaron metabolitos importantes de empagliflozina en el plasma humano, tal como se define mediante al menos el 10 % del material total relacionado con el fármaco, y los metabolitos más abundantes fueron tres conjugados glucurónidos (2-, 3- y 6-O glucurónido). Los estudios in vitro sugirieron que la principal vía metabólica de empagliflozina en humanos es la glucuronidación por las uridina 5’-difosfo-glucuronosiltransferasas UGT2B7, UGT1A3, UGT1A8 y UGT1A9.
Eliminación
De acuerdo al análisis farmacocinético poblacional, se calculó que la semivida de eliminación terminal aparente de empagliflozina era de 12,4 horas y que el aclaramiento oral aparente era de 10,6 l/hora.
Las variabilidades interindividual y residual para el aclaramiento oral de empagliflozina fueron del 39,1 % y del 35,8 %, respectivamente. Con una pauta posológica de una vez al día, las concentraciones plasmáticas de empagliflozina en estado estacionario se alcanzaron en la quinta dosis. Acorde con la semivida, en el estado estacionario se observó una acumulación de hasta el 22 % con respecto al AUC plasmática. Tras la administración de una solución oral de [14C]-empagliflozina a voluntarios sanos, aproximadamente el 96 % de la radioactividad relacionada con el fármaco se eliminó por las heces (41 %) o la orina (54 %). La mayor parte de la radioactividad relacionada con el fármaco que se recuperó en las heces fue el fármaco original sin cambios y aproximadamente la mitad de la radioactividad relacionada con el fármaco excretado por la orina fue el fármaco original sin cambios.
Poblaciones especiales
Insuficiencia renal
En pacientes con insuficiencia renal leve, moderada o grave (aclaramiento de creatinina < 30 - < 90 ml/min) y pacientes con fallo renal/enfermedad renal terminal (ERT), el AUC de empagliflozina aumentó en aproximadamente el 18 %, 20 %, 66 % y 48 % respectivamente en comparación con los individuos con una función renal normal. Los niveles plasmáticos máximos de empagliflozina fueron similares en los sujetos con insuficiencia renal moderada y fallo renal/ERT en comparación con los pacientes con una función renal normal. Los niveles plasmáticos máximos de empagliflozina fueron aproximadamente un 20 % más altos en los sujetos con insuficiencia renal leve y grave en comparación con los sujetos con una función renal normal. El análisis farmacocinético poblacional demostró que el aclaramiento oral aparente de empagliflozina disminuía con un descenso en el aclaramiento de creatinina, dando lugar a un aumento en la exposición al fármaco.
Insuficiencia hepática
En sujetos con insuficiencia hepática leve, moderada y grave según la clasificación Child-Pugh, el AUC de empagliflozina aumentó en aproximadamente el 23 %, 47 %, y 75 % y la Cmax aumentó en el 4 %, 23 % y 48 % respectivamente en comparación con los sujetos con función hepática normal.
Índice de masa corporal
El índice de masa corporal no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de empagliflozina según un análisis farmacocinético poblacional. En este análisis, se estimó que el AUC era un 5,82 %, 10,4 % y 17,3 % inferior en sujetos con un IMC de 30, 35 y 45 kg/m2 respectivamente, en comparación con sujetos con un índice de masa corporal de 25 kg/m2.
Sexo
El sexo no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de empagliflozina según un análisis farmacocinético poblacional.
Raza
En el análisis farmacocinético poblacional, se estimó que el AUC era un 13,5 % más alta en asiáticos con un índice de masa corporal de 25 kg/m2 en comparación con los no asiáticos con un índice de masa corporal de 25 kg/m2.
Pacientes de edad avanzada
La edad no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de empagliflozina según un análisis farmacocinético poblacional.
Población pediátrica
Un ensayo pediátrico de fase I examinó la farmacocinética y la farmacodinámica de empagliflozina (5 mg, 10 mg y 25 mg) en niños y adolescentes de ≥ 10 a < 18 años de edad con diabetes mellitus tipo 2. Las respuestas farmacocinéticas y farmacodinámicas observadas fueron consistentes con las obtenidas en sujetos adultos.
Un estudio pediátrico de fase III examinó la farmacocinética y la farmacodinámica (cambio de la HbA1c desde el momento inicial) de empagliflozina en dosis de 10 mg con un posible aumento de dosis a 25 mg en niños y adolescentes de entre 10 y 17 años de edad con diabetes mellitus tipo 2. La relación exposición-respuesta observada fue en conjunto comparable en adultos y en niños y adolescentes. La administración oral de empagliflozina dio lugar a una exposición dentro del intervalo observado en pacientes adultos.
La media geométrica de las concentraciones mínimas y la media geométrica de las concentraciones observadas 1,5 horas después de la administración en estado estacionario fueron de 26,6 nmol/l y 308 nmol/l con empagliflozina en dosis de 10 mg una vez al día y de 67,0 nmol/l y 525 nmol/l con empagliflozina en dosis de 25 mg una vez al día.
Metformina
Absorción
Después de una dosis oral de metformina, el tmax se alcanza en 2,5 horas. La biodisponibilidad absoluta de un comprimido de 500 mg u 850 mg de hidrocloruro de metformina es de aproximadamente el 50-60 % en sujetos sanos. Después de una dosis oral, la fracción no absorbida recuperada en las heces fue del 20-30 %. Tras la administración oral, la absorción de metformina es saturable e incompleta. Se supone que la farmacocinética de la absorción de metformina no es lineal. A las dosis y pautas posológicas recomendadas de metformina, las concentraciones plasmáticas en estado estacionario se alcanzan en un plazo de 24 a 48 horas y son generalmente inferiores a 1 microgramo/ml. En ensayos clínicos controlados, las concentraciones plasmáticas máximas de metformina (Cmax) no sobrepasaron los 5 microgramos/ml, ni siquiera a las dosis máximas.
Los alimentos retrasan ligeramente y disminuyen el grado de absorción de metformina. Tras la administración de una dosis de 850 mg de hidrocloruro de metformina, se observó una concentración plasmática máxima un 40 % menor, una reducción del 25 % en el AUC (área bajo la curva) y una prolongación de 35 minutos en el tiempo transcurrido hasta alcanzar la concentración plasmática máxima. Se desconoce la relevancia clínica de esta reducción.
Distribución
La unión a proteínas plasmáticas es insignificante. Metformina se difunde por los eritrocitos. La concentración sanguínea máxima es menor que la concentración plasmática máxima y aparece aproximadamente al mismo tiempo. Los glóbulos rojos probablemente representan un compartimento secundario de distribución. El volumen de distribución (Vd) medio osciló entre 63 y 276 l.
Biotransformación
Metformina se excreta inalterada en la orina. No se han identificado metabolitos en humanos.
Eliminación
El aclaramiento renal de metformina es > 400 ml/min, lo que indica que metformina se elimina mediante filtración glomerular y secreción tubular. Después de una dosis oral, la semivida de eliminación terminal aparente es de aproximadamente 6,5 horas.
Cuando la función renal está afectada, el aclaramiento renal disminuye en proporción al de la creatinina y, por tanto, la semivida de eliminación se prolonga, lo que da lugar a un aumento de los niveles de metformina en plasma.
Poblaciones especiales
Población pediátrica
Estudio de dosis única: tras dosis únicas de 500 mg de hidrocloruro de metformina los pacientes pediátricos han mostrado un perfil farmacocinético similar al observado en adultos sanos.
Estudio de dosis múltiples: tras dosis repetidas de 500 mg dos veces al día durante 7 días en pacientes pediátricos, la concentración plasmática máxima (Cmax) y la exposición sistémica (AUC0-t) se redujeron en aproximadamente un 33 % y un 40 %, respectivamente, en comparación con adultos diabéticos que recibieron dosis repetidas de 500 mg dos veces al día durante 14 días. Puesto que la dosis se ajusta individualmente en base al control glucémico, este hecho posee relevancia clínica limitada.
5.3 - Datos preclínicos sobre seguridad de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Empagliflozina y metformina
Se realizaron estudios de toxicidad general en ratas durante un máximo de 13 semanas con la combinación de empagliflozina y metformina y no se observaron órganos diana adicionales cuando se comparó con empagliflozina o metformina en monoterapia. Algunas respuestas aumentaron con el tratamiento combinado, como efectos en la fisiología renal, el equilibrio electrolítico y el estado ácido-base. No obstante, solo la hipocloremia se consideró adversa a exposiciones de aproximadamente 9 y 3 veces la exposición clínica del AUC a la dosis máxima recomendada de empagliflozina y metformina, respectivamente.
Un estudio de desarrollo embriofetal en ratas embarazadas no indicó efecto teratogénico atribuido a la administración conjunta de empagliflozina y metformina a exposiciones de aproximadamente 14 veces la exposición clínica del AUC de empagliflozina asociada a la dosis más alta, y 4 veces la exposición clínica del AUC de metformina asociada a la dosis de 2 000 mg.
Empagliflozina
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, genotoxicidad, fertilidad y desarrollo embrionario temprano.
En estudios de toxicidad a largo plazo con roedores y perros, se observaron signos de toxicidad a exposiciones iguales o superiores a 10 veces la dosis clínica de empagliflozina. La mayor parte de las toxicidades fueron compatibles con la farmacología secundaria relacionada con pérdida de glucosa por la orina y desequilibrios electrolíticos, incluida la disminución del peso y la grasa corporales, aumento del consumo de alimentos, diarrea, deshidratación, disminución de los niveles de glucosa sérica y aumentos en otros parámetros séricos que reflejan el aumento del metabolismo de las proteínas y la gluconeogénesis, cambios urinarios como la poliuria y la glucosuria y cambios microscópicos, incluida la mineralización en el riñón y en algunos tejidos blandos y vasculares. Las evidencias microscópicas de los efectos de una farmacología exagerada en el riñón que se observaron en algunas especies incluyeron dilatación tubular y mineralización tubular y pélvica a aproximadamente 4 veces la exposición del AUC clínica de empagliflozina asociada a la dosis de 25 mg.
Empagliflozina no es genotóxica.
En un estudio de carcinogénesis de 2 años de duración, empagliflozina no aumentó la incidencia de tumores en ratas hembra hasta la dosis máxima de 700 mg/kg/día, lo que corresponde a aproximadamente 72 veces la exposición clínica máxima del AUC a empagliflozina. En las ratas macho, a las dosis más altas se observaron lesiones proliferativas vasculares benignas relacionadas con el tratamiento (hemangiomas) del ganglio linfático mesentérico, pero no a la dosis de 300 mg/kg/día, lo que corresponde a aproximadamente 26 veces la exposición clínica máxima a empagliflozina. Se observaron tumores celulares intersticiales en los testículos con una mayor incidencia en ratas a 300 mg/kg/día o más, pero no a 100 mg/kg/día, lo que corresponde a aproximadamente 18 veces la exposición clínica máxima a empagliflozina. Ambos tumores son frecuentes en ratas, pero es improbable que sean relevantes en los humanos.
Empagliflozina no aumentó la incidencia de tumores en ratones hembra a dosis de hasta 1 000 mg/kg/día, lo que corresponde a aproximadamente 62 veces la exposición clínica máxima a empagliflozina. Empagliflozina indujo tumores renales en ratones macho a dosis de 1 000 mg/kg/día, pero no a la dosis de 300 mg/kg/día, lo que corresponde a aproximadamente 11 veces la exposición clínica máxima a empagliflozina. El modo de acción de estos tumores depende de la predisposición natural del ratón macho a presentar una patología renal y una vía metabólica que no refleja la de los humanos. Los tumores renales de los ratones macho no se consideran relevantes para los humanos.
A exposiciones suficientemente superiores a la exposición en humanos después de dosis terapéuticas, empagliflozina no tuvo efectos adversos sobre la fertilidad o el desarrollo embrionario temprano. La administración de empagliflozina durante el periodo de organogénesis no fue teratogénica. Solo a dosis tóxicas para la madre, empagliflozina también provocó que los huesos de las extremidades de la rata se doblasen, así como un aumento de la muerte embriofetal en el conejo.
En estudios de la toxicidad prenatal y posnatal en ratas, se observó una reducción en el aumento de peso de la descendencia a exposiciones maternas de aproximadamente 4 veces la exposición clínica máxima a empagliflozina. Dicho efecto no se observó a la exposición sistémica igual a la máxima exposición clínica a empagliflozina. La relevancia de este hallazgo para los humanos no está clara.
En un estudio de toxicidad juvenil en ratas, cuando se administró empagliflozina desde el día 21 posnatal hasta el día 90 posnatal, se observó una dilatación pélvica y de los túbulos renales, no adversa y de mínima a leve, en ratas jóvenes solo a 100 mg/kg/día, lo que se aproxima a 11 veces la dosis clínica máxima de 25 mg. Estos hallazgos no estaban presentes tras un periodo de recuperación de 13 semanas sin fármaco.
Metformina
Los datos de los estudios preclínicos para metformina no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad o potencial carcinogénico o toxicidad para la reproducción. A niveles de dosis de 500 mg/kg/día administrados a ratas Wistar Hannover, asociados a 7 veces la dosis humana máxima recomendada de metformina, se observó teratogénesis de metformina, que se manifestó principalmente como un aumento del número de malformaciones esqueléticas.
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Synjardy 5 mg/850 mg comprimidos recubiertos con película y Synjardy 5 mg/1.000 mg comprimidos recubiertos con película
Núcleo del comprimido
Almidón de maíz
Copovidona (valor K nominalmente 28)
Sílice coloidal anhidra
Estearato de magnesio
Cubierta pelicular
Hipromelosa
Macrogol 400
Dióxido de titanio (E171)
Talco
Óxido de hierro amarillo (E172)
Synjardy 12,5 mg/850 mg comprimidos recubiertos con película y Synjardy 12,5 mg/1.000 mg comprimidos recubiertos con película
Núcleo del comprimido
Almidón de maíz
Copovidona (valor K nominalmente 28)
Sílice coloidal anhidra
Estearato de magnesio
Cubierta pelicular
Hipromelosa
Macrogol 400
Dióxido de titanio (E171)
Talco
Óxido de hierro negro (E172)
Óxido de hierro rojo (E172)
6.2 - Incompatibilidades de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
No procede.
6.3 - Período de validez de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
3 años
6.4 - Precauciones especiales de conservación de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
No requiere condiciones especiales de conservación.
6.5 - Naturaleza y contenido del recipiente de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
Blísteres unidosis perforados de PVC/PVDC/aluminio.
Tamaños de envase de 10 x 1, 14 x 1, 30 x 1, 56 x 1, 60 x 1, 90 x 1 y 100 x 1 comprimidos recubiertos con película y envases múltiples que contienen 120 (2 envases de 60 x 1), 180 (2 envases de 90 x 1) y 200 (2 envases de 100 x 1) comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de SYNJARDY 12,5 mg/1000 mg Comp. recub. con película
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Boehringer Ingelheim International GmbH
Binger Str. 173
55216 Ingelheim am Rhein
Alemania
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Synjardy 5 mg/850 mg comprimidos recubiertos con película
EU/1/15/1003/001
EU/1/15/1003/002
EU/1/15/1003/003
EU/1/15/1003/004
EU/1/15/1003/005
EU/1/15/1003/037
EU/1/15/1003/006
EU/1/15/1003/007
EU/1/15/1003/008
EU/1/15/1003/009
Synjardy 5 mg/1.000 mg comprimidos recubiertos con película
EU/1/15/1003/010
EU/1/15/1003/011
EU/1/15/1003/012
EU/1/15/1003/013
EU/1/15/1003/014
EU/1/15/1003/038
EU/1/15/1003/015
EU/1/15/1003/016
EU/1/15/1003/017
EU/1/15/1003/018
Synjardy 12,5 mg/850 mg comprimidos recubiertos con película
EU/1/15/1003/019
EU/1/15/1003/020
EU/1/15/1003/021
EU/1/15/1003/022
EU/1/15/1003/023
EU/1/15/1003/039
EU/1/15/1003/024
EU/1/15/1003/025
EU/1/15/1003/026
EU/1/15/1003/027
Synjardy 12,5 mg/1.000 mg comprimidos recubiertos con película
EU/1/15/1003/028
EU/1/15/1003/029
EU/1/15/1003/030
EU/1/15/1003/031
EU/1/15/1003/032
EU/1/15/1003/040
EU/1/15/1003/033
EU/1/15/1003/034
EU/1/15/1003/035
EU/1/15/1003/036
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 27/mayo/2015
Fecha de la última renovación: 01/abril/2020
10. - FECHA DE LA REVISIÓN DEL TEXTO
04/noviembre/2024
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos https://www.ema.europa.eu.

