 LORVIQUA 100 MG COMPRIMIDOS RECUBIERTOS CON PELÍCULA
LORVIQUA 100 MG COMPRIMIDOS RECUBIERTOS CON PELÍCULA

| ATC: Lorlatinib |
| PA: Lorlatinib |
| EXC: Lactosa monohidrato y otros. |
Envases
- Env. con 30
- DHSC: Medicamento de diagnóstico hospitalario sin cupón precinto
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica- Fi: Medicamento incluido en la financiación del SNS
- Facturable SNS: NO
- Comercializado: Si
- Situación: Alta
- Código Nacional: 725704
- EAN13: 8470007257042
- Conservar en frío: No
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
LORVIQUA 100 mg Comp. recub. con película
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Lorviqua 25 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene 25 mg de lorlatinib.
Excipiente con efecto conocido
Cada comprimido recubierto con película contiene 1,58 mg de lactosa monohidrato.
Lorviqua 100 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene 100 mg de lorlatinib.
Excipiente con efecto conocido
Cada comprimido recubierto con película contiene 4,20 mg de lactosa monohidrato.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. - FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Lorviqua 25 mg comprimidos recubiertos con película
Comprimido recubierto con película de liberación inmediata de color rosa claro y redondo (8 mm), con “Pfizer” grabado en una cara y “25” y “LLN” en la otra.
Lorviqua 100 mg comprimidos recubiertos con película
Comprimido recubierto con película de liberación inmediata de color rosa oscuro y ovalado (8,5 × 17 mm), con “Pfizer” grabado en una cara y “LLN 100” en la otra.
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de LORVIQUA 100 mg Comp. recub. con película
Lorviqua en monoterapia está indicado para el tratamiento de pacientes adultos con cáncer de pulmón no microcítico (CPNM) avanzado positivo para la quinasa del linfoma anaplásico (ALK) no tratado previamente con un inhibidor de ALK.
Lorviqua en monoterapia está indicado para el tratamiento de pacientes adultos con CPNM avanzado positivo para ALK cuya enfermedad ha progresado tras recibir:
- alectinib o ceritinib como primer tratamiento con un inhibidor de la tirosina quinasa (TKI) ALK; o
- crizotinib y al menos otro TKI ALK.
4.2 - Posología y administración de LORVIQUA 100 mg Comp. recub. con película
4.3 - Contraindicaciones de LORVIQUA 100 mg Comp. recub. con película
Hipersensibilidad a lorlatinib o a alguno de los excipientes incluidos en la sección 6.1.
Uso concomitante de inductores potentes del CYP3A4/5 (ver las secciones 4.4 y 4.5).
4.4 - Advertencias y Precauciones de LORVIQUA 100 mg Comp. recub. con película
Hiperlipidemia
El uso de lorlatinib se ha relacionado con aumentos en los niveles de colesterol y triglicéridos séricos (ver sección 4.8). La mediana de tiempo hasta la aparición de aumento grave de los niveles de colesterol y triglicéridos séricos es de 104 días (rango de 29 a 518 días) y 120 días (rango de 15 a 780 días), respectivamente. Se deben monitorizar los niveles de colesterol y triglicéridos séricos antes del inicio del tratamiento con lorlatinib; 2, 4 y 8 semanas después de iniciar el tratamiento con lorlatinib y regularmente a partir de entonces. Inicie o aumente la dosis de los hipolipemiantes, si está indicado (ver sección 4.2).
Efectos sobre el sistema nervioso central
Se han observado efectos sobre el sistema nervioso central (SNC) en pacientes en tratamiento con lorlatinib, incluidos efectos psicóticos y cambios en la función cognitiva, el estado de ánimo, el estado mental o el habla (ver sección 4.8). Es posible que se requiera la modificación o la interrupción de la dosis en aquellos pacientes que presenten efectos sobre el SNC (ver sección 4.2).
Bloqueo auriculoventricular
Lorlatinib se estudió en una población de pacientes que excluyó a aquellos con bloqueo AV de segundo o tercer grado (a menos que presentaran electroestimulación) o con cualquier bloqueo AV con intervalo PR >220 ms. Se ha notificado prolongación del intervalo PR y bloqueo AV en pacientes tratados con lorlatinib (ver sección 5.2). Monitorice el electrocardiograma (ECG) antes de iniciar el tratamiento con lorlatinib y mensualmente a partir de entonces, especialmente en pacientes con afecciones que predispongan a la aparición de acontecimientos cardíacos clínicamente significativos. Puede ser necesaria una modificación de la dosis en aquellos pacientes que presentan bloqueo AV (ver sección 4.2).
Disminución de la fracción de eyección del ventrículo izquierdo
Se ha notificado una disminución de la fracción de eyección del ventrículo izquierdo (FEVI) en pacientes tratados con lorlatinib que tenían una evaluación al inicio del estudio y al menos una evaluación de seguimiento de la FEVI. Según los datos de los estudios clínicos disponibles, no es posible determinar una relación causal entre los efectos sobre los cambios en la contractilidad cardíaca y lorlatinib. En pacientes con factores de riesgo cardíaco y aquellos con afecciones que pueden afectar a la FEVI, se debe considerar una monitorización cardíaca, incluida la evaluación de la FEVI al inicio y durante el tratamiento. En pacientes que presenten signos/síntomas cardíacos relevantes durante el tratamiento, se debe considerar una monitorización cardíaca, incluida la evaluación de la FEVI.
Aumento de los niveles de lipasa y amilasa
Se han producido aumentos de los niveles de lipasa y/o amilasa en pacientes que recibían lorlatinib (ver sección 4.8). La mediana de tiempo hasta la aparición del aumento de los niveles de lipasa y amilasa sérica es de 141 días (rango de 1 a 1 091 días) y 138 días (rango de 1 a 1 112 días), respectivamente. Se debe considerar el riesgo de pancreatitis en pacientes que reciben lorlatinib debido a una hipertrigliceridemia concomitante y/o un posible mecanismo intrínseco. Los pacientes deben ser controlados para detectar aumentos de los niveles de lipasa y amilasa antes del inicio del tratamiento con lorlatinib y posteriormente, de forma regular, según se indique clínicamente (ver sección 4.2).
Enfermedad pulmonar intersticial/neumonitis
Se han producido reacciones adversas pulmonares graves o potencialmente mortales compatibles con EPI/neumonitis con lorlatinib (ver sección 4.8). Se debe evaluar inmediatamente a cualquier paciente que presente un empeoramiento de los síntomas respiratorios indicativos de EPI/neumonitis (por ejemplo, disnea, tos y fiebre) para detectar EPI/neumonitis. El tratamiento con lorlatinib se debe interrumpir y/o suspender permanentemente según la gravedad (ver sección 4.2).
Hipertensión arterial
Se ha notificado hipertensión arterial en pacientes tratados con lorlatinib (ver sección 4.8). Se debe vigilar la tensión arterial antes de iniciar el tratamiento con lorlatinib. La tensión arterial se debe controlar después de 2 semanas y al menos una vez al mes durante el tratamiento con lorlatinib. Se debe interrumpir el uso de lorlatinib y reanudarlo con una dosis reducida o suspenderlo permanentemente en función de la gravedad (ver sección 4.2).
Hiperglucemia
Se ha producido hiperglucemia en pacientes tratados con lorlatinib (ver sección 4.8). Se debe evaluar la glucosa sérica en ayunas antes de iniciar el tratamiento con lorlatinib y, posteriormente, se debe controlar periódicamente de acuerdo con las recomendaciones nacionales. Se debe suspender el uso de lorlatinib y reanudarlo con una dosis reducida o suspenderlo permanentemente en función de la gravedad (ver sección 4.2).
Interacciones farmacológicas
En un estudio realizado en voluntarios sanos, el uso concomitante de lorlatinib y rifampicina, un potente inductor del CYP3A4/5, se relacionó con aumentos de los niveles de alanina aminotransferasa (ALT) y aspartato aminotransferasa (AST) sin aumento de los niveles de bilirrubina total y fosfatasa alcalina (ver sección 4.5). El uso concomitante de un inductor potente del CYP3A4/5 está contraindicado (ver las secciones 4.3 y 4.5). No se observaron cambios clínicamente significativos en las pruebas de función hepática en sujetos sanos después de recibir una combinación de lorlatinib con el inductor moderado del CYP3A4/5, modafinilo (ver sección 4.5).
Se debe evitar la administración simultánea de lorlatinib con sustratos del CYP3A4/5 con índices terapéuticos estrechos, incluidos entre otros, alfentanilo, ciclosporina, dihidroergotamina, ergotamina, fentanilo, anticonceptivos hormonales, pimozida, quinidina, sirolimus y tacrolimus, ya que lorlatinib puede reducir la concentración de estos medicamentos (ver sección 4.5).
Fertilidad y embarazo
Durante el tratamiento con lorlatinib y durante al menos 14 semanas tras la dosis final, los pacientes varones con parejas femeninas en edad fértil deben usar métodos anticonceptivos efectivos, incluyendo un preservativo, y los pacientes varones con parejas embarazadas deben usar preservativos (ver sección 4.6). La fertilidad masculina puede verse comprometida con el tratamiento con lorlatinib (ver sección 5.3). Los hombres deben solicitar asesoramiento sobre la preservación efectiva de su fertilidad antes del tratamiento. A las mujeres en edad fértil se les debe recomendar que eviten quedarse embarazadas durante el tratamiento con lorlatinib. Se requiere un método anticonceptivo no hormonal altamente efectivo para las mujeres durante el tratamiento con lorlatinib, puesto que lorlatinib puede anular la eficacia de los anticonceptivos hormonales (ver las secciones 4.5 y 4.6). Si el uso de un método anticonceptivo hormonal es inevitable, entonces se debe usar un preservativo en combinación con el método hormonal. Se debe continuar el uso de anticonceptivos efectivos durante al menos 35 días tras finalizar el tratamiento (ver sección 4.6). Se desconoce si lorlatinib afecta a la fertilidad femenina.
Intolerancia a la lactosa
Este medicamento contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
Sodio en dietas
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por comprimido de 25 mg o 100 mg. Se debe informar a los pacientes con dietas bajas en sodio que este medicamento se considera esencialmente “exento de sodio”.
4.5 - Interacciones con otros medicamentos de LORVIQUA 100 mg Comp. recub. con película
Interacciones farmacocinéticas
Los datos in vitro indican que lorlatinib se metaboliza principalmente por el CYP3A4 y la uridinadifosfato glucuroniltransferasa (UGT)1A4, y en menor medida por el CYP2C8, CYP2C19, CYP3A5 y la UGT1A3.
Efecto de otros medicamentos sobre lorlatinib
Inductores del CYP3A4/5
La rifampicina, un potente inductor del CYP3A4/5, administrada a dosis orales de 600 mg una vez al día durante 12 días, redujo el área bajo la curva (AUCinf, por sus siglas en inglés) media de lorlatinib en un 85% y la Cmáx en un 76% de una dosis oral única de 100 mg de lorlatinib en voluntarios sanos; también se observaron aumentos en la AST y la ALT. La administración concomitante de lorlatinib con inductores potentes del CYP3A4/5 (por ejemplo, rifampicina, carbamazepina, enzalutamida, mitotano, fenitoína y la hierba de San Juan) puede disminuir las concentraciones plasmáticas de lorlatinib. El uso de un inductor potente del CYP3A4/5 con lorlatinib está contraindicado (ver las secciones 4.3 y 4.4). No se observaron cambios clínicamente significativos en los resultados de las pruebas de función hepática después de la administración de una combinación de una dosis oral única de 100 mg de lorlatinib con el inductor moderado del CYP3A4/5, modafinilo (400 mg una vez al día durante 19 días) en voluntarios sanos. El uso concomitante de modafinilo no tuvo un efecto clínicamente significativo sobre la farmacocinética de lorlatinib.
Inhibidores del CYP3A4/5
Itraconazol, un potente inhibidor del CYP3A4/5, administrado a dosis orales de 200 mg una vez al día durante 5 días, aumentó el AUCinf media de lorlatinib en un 42% y la Cmáx en un 24% de una dosis oral única de 100 mg de lorlatinib en voluntarios sanos. La administración concomitante de lorlatinib con inhibidores potentes del CYP3A4/5 (por ejemplo, boceprevir, cobicistat, itraconazol, ketoconazol, posaconazol, troleandomicina, voriconazol, ritonavir y paritaprevir en combinación con ritonavir y ombitasvir y/o dasabuvir, y ritonavir en combinación con elvitegravir, indinavir, lopinavir o tipranavir) puede aumentar las concentraciones plasmáticas de lorlatinib. Los productos con pomelo también pueden aumentar las concentraciones plasmáticas de lorlatinib y deben evitarse. Se debe considerar la administración de un medicamento concomitante alternativo con un menor potencial para inhibir el CYP3A4/5. Si se debe administrar de forma concomitante un inhibidor potente del CYP3A4/5, se recomienda reducir la dosis de lorlatinib (ver sección 4.2).
Efecto de lorlatinib sobre otros medicamentos
Sustratos del CYP3A4/5
Los estudios in vitro indicaron que lorlatinib es un inhibidor dependiente del tiempo, así como un inductor del CYP3A4/5. Lorlatinib 150 mg administrado por vía oral una vez al día durante 15 días disminuyó el AUCinf y la Cmáx de una dosis oral única de 2 mg de midazolam (un sustrato sensible del CYP3A) en un 61% y un 50%, respectivamente; por lo tanto, lorlatinib es un inductor moderado del CYP3A. Por consiguiente, se debe evitar la administración concomitante de lorlatinib con sustratos del CYP3A4/5 con índices terapéuticos estrechos, incluidos, entre otros, alfentanilo, ciclosporina, dihidroergotamina, ergotamina, fentanilo, anticonceptivos hormonales, pimozida, quinidina, sirolimus y tacrolimus, ya que lorlatinib puede reducir la concentración de estos medicamentos (ver sección 4.4).
Sustratos del CYP2B6
Lorlatinib 100 mg una vez al día durante 15 días disminuyó el AUCinf y la Cmáx de una dosis oral única de 100 mg de bupropión (un sustrato combinado del CYP2B6 y CYP3A4) en un 49,5% y un 53%, respectivamente. Por consiguiente, lorlatinib es un inductor débil del CYP2B6, y no es necesario ajustar la dosis cuando se usa lorlatinib en combinación con medicamentos que se metabolizan principalmente por el CYP2B6.
Sustratos del CYP2C9
Lorlatinib 100 mg una vez al día durante 15 días disminuyó el AUCinf y la Cmáx de una dosis oral única de 500 mg de tolbutamida (un sustrato sensible del CYP2C9) en un 43% y un 15%, respectivamente. Por consiguiente, lorlatinib es un inductor débil del CYP2C9, y no es necesario ajustar la dosis de los medicamentos que se metabolizan principalmente por el CYP2C9. Sin embargo, se debe monitorizar a los pacientes en caso de tratamiento concomitante con medicamentos con margen terapéutico estrecho metabolizados por el CYP2C9 (por ejemplo, anticoagulantes cumarínicos).
Sustratos de la UGT
Lorlatinib 100 mg una vez al día durante 15 días disminuyó el AUCinf y la Cmáx de una dosis oral única de 500 mg de acetaminofeno (también conocido como paracetamol) (un sustrato de la UGT, SULT y el CYP1A2, 2A6, 2D6 y 3A4) en un 45% y un 28%, respectivamente. Por consiguiente, lorlatinib es un inductor débil de la UGT, y no es necesario ajustar la dosis de los medicamentos que se metabolizan principalmente por la UGT. Sin embargo, se debe controlar a los pacientes en caso de tratamiento concomitante con medicamentos con margen terapéutico estrecho metabolizados por la UGT.
Sustratos de la glucoproteína P
Lorlatinib 100 mg una vez al día durante 15 días disminuyó el AUCinf y la Cmáx de una dosis oral única de 60 mg de fexofenadina (un sustrato sensible de la glucoproteína P [P-gp]) en un 67% y un 63%, respectivamente. Por consiguiente, lorlatinib es un inductor moderado de la P-gp. Los medicamentos que son sustratos de la P-gp con margen terapéutico estrecho (por ejemplo, digoxina, dabigatrán etexilato) se deben usar con precaución en combinación con lorlatinib debido a la probabilidad de reducir las concentraciones plasmáticas de estos sustratos.
Estudios in vitro de inhibición e inducción de otras enzimas CYP
In vitro, lorlatinib tiene un bajo potencial de causar interacciones farmacológicas por inducción del CYP1A2.
Estudios in vitro con transportadores de fármacos diferentes a la P-gp
Los estudios in vitro indicaron que lorlatinib puede tener el potencial de inhibir la BCRP (tracto gastrointestinal), OATP1B1, OATP1B3, OCT1, MATE1 y OAT3 a concentraciones clínicamente relevantes. Lorlatinib se debe utilizar con precaución en combinación con sustratos de la BCRP, OATP1B1, OATP1B3, OCT1, MATE1 y OAT3, ya que no se pueden descartar cambios clínicamente relevantes en la exposición plasmática de estos sustratos.
4.6 - Embarazo y Lactancia de LORVIQUA 100 mg Comp. recub. con película
Mujeres en edad fértil/Anticoncepción en varones y mujeres
A las mujeres en edad fértil se les debe recomendar que eviten quedarse embarazadas durante el tratamiento con lorlatinib. Se requiere un método anticonceptivo no hormonal altamente efectivo para las mujeres durante el tratamiento con lorlatinib, puesto que lorlatinib puede anular la eficacia de los anticonceptivos hormonales (ver las secciones 4.4 y 4.5). Si el uso de un método anticonceptivo hormonal es inevitable, entonces se debe usar un preservativo en combinación con el método hormonal. Se debe continuar el uso de anticonceptivos efectivos durante al menos 35 días tras finalizar el tratamiento.
Durante el tratamiento con lorlatinib y durante al menos 14 semanas tras la dosis final, los pacientes varones con parejas femeninas en edad fértil deben usar métodos anticonceptivos efectivos, incluyendo un preservativo, y los pacientes varones con parejas embarazadas deben usar preservativos.
Embarazo
Los estudios realizados en animales han mostrado toxicidad embriofetal (ver sección 5.3). No hay datos relativos al uso de lorlatinib en mujeres embarazadas. Lorlatinib puede causar daño fetal cuando se administra a una mujer embarazada.
No se recomienda utilizar lorlatinib durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
Se desconoce si lorlatinib y sus metabolitos se excretan en la leche materna. No se puede excluir el riesgo en recién nacidos/lactantes.
Lorlatinib no debe utilizarse durante la lactancia. Debe interrumpirse la lactancia durante el tratamiento con lorlatinib y durante los 7 días siguientes tras recibir la dosis final.
Fertilidad
Según los hallazgos de seguridad no clínicos, la fertilidad masculina puede verse comprometida con el tratamiento con lorlatinib (ver sección 5.3). Se desconoce si lorlatinib afecta a la fertilidad femenina. Los hombres deben solicitar asesoramiento para la preservación efectiva de su fertilidad antes del tratamiento.
4.7 - Efectos sobre la capacidad de conducción de LORVIQUA 100 mg Comp. recub. con película
La influencia de lorlatinib sobre la capacidad para conducir y utilizar máquinas es moderada. Se debe tener precaución al conducir o utilizar máquinas ya que los pacientes pueden experimentar efectos sobre el SNC (ver sección 4.8).
4.8 - Reacciones Adversas de LORVIQUA 100 mg Comp. recub. con película
Resumen del perfil de seguridad
Las reacciones adversas notificadas con más frecuencia fueron hipercolesterolemia (81,1%), hipertrigliceridemia (67,2%), edema (55,7%), neuropatía periférica (43,7%), aumento de peso (30,9%), efectos cognitivos (27,7%), cansancio (27,3%), artralgia (23,5%), diarrea (22,9%) y efectos sobre el estado de ánimo (21,0%).
Se notificaron reacciones adversas graves en el 7,4% de los pacientes que recibieron lorlatinib. Las reacciones adversas graves más frecuentes fueron los efectos cognitivos y la neumonitis.
Las reducciones de dosis debidas a reacciones adversas se produjeron en el 20,0% de los pacientes que recibieron lorlatinib. Las reacciones adversas más frecuentes que condujeron a la reducción de la dosis fueron edema y neuropatía periférica. La interrupción permanente del tratamiento relacionada con reacciones adversas se produjo en el 3,2% de los pacientes que recibieron lorlatinib. Las reacciones adversas más frecuentes que condujeron a interrupciones permanentes fueron los efectos cognitivos, la neuropatía periférica, la neumonitis y los efectos psicóticos.
Tabla de reacciones adversas
En la tabla 2 se presentan las reacciones adversas que se produjeron en 476 pacientes adultos tratados con 100 mg de lorlatinib una vez al día con CPNM avanzado del estudio A (N = 327) y del estudio CROWN (N = 149).
Las reacciones adversas enumeradas en la tabla 2 se presentan según el sistema de clasificación de órganos y por categorías de frecuencia, definidas según la siguiente convención: muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes (≥1/1 000 a <1/100), raras (≥1/10 000 a <1/1 000) o muy raras (<1/10 000). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 2. Reacciones adversas
| Clasificación por órganos y sistemas y reacción adversa | Categoría de frecuencia | Todos los grados % | Grados 3-4 % |
| Anemia | Muy frecuentes | 18,5 | 4,2 |
| Hiperglucemia | Frecuentes | 9,2 | 3,2 |
| Cambios en el estado mental | Frecuentes | 2,0 | 1,7 |
| Efectos sobre el hablag | Frecuentes | 8,2 | 0,6 |
| Trastorno de la visiónh | Muy frecuentes | 17,2 | 0,2 |
| Hipertensión arterial | Muy frecuentes | 13,0 | 6,1 |
| Neumonitisi | Frecuentes | 1,9 | 0,6 |
| Estreñimiento | Muy frecuentes | 17,4 | 0,2 |
| Erupciónj | Muy frecuentes | 13,7 | 0,2 |
| Mialgiak | Muy frecuentes | 19,3 | 0,2 |
| Cansanciom | Muy frecuentes | 27,3 | 1,3 |
| Prolongación del intervalo PR en el electrocardiograma | Poco frecuentes | 0,8 | 0 |
Las reacciones adversas que representan el mismo concepto médico o afección fueron agrupadas y se notificaron como una única reacción adversa en la tabla anterior. Los términos realmente notificados en los estudios y que contribuyen a la reacción adversa relevante se indican entre paréntesis, tal y como se detalla a continuación.
a Hipercolesterolemia (incluye colesterol sanguíneo elevado, hipercolesterolemia).
b Hipertrigliceridemia (incluye triglicéridos sanguíneos elevados, hipertrigliceridemia).
c Efectos sobre el estado de ánimo (incluye trastorno afectivo, inestabilidad afectiva, agresividad, nerviosismo, irritabilidad, ansiedad, trastorno bipolar de tipo I, estado de ánimo deprimido, depresión, síntomas depresivos, estado de ánimo eufórico, irritabilidad, manía, estado de ánimo alterado, cambios de humor, crisis de angustia, cambio de personalidad, estrés).
d Efectos psicóticos (incluye alucinaciones auditivas, alucinaciones, alucinaciones visuales).
e Efectos cognitivos (incluye acontecimientos encuadrados en el epígrafe de trastornos del sistema nervioso según el sistema de clasificación de órganos: amnesia, trastorno cognitivo, demencia, alteración de la atención, deterioro de la memoria, deterioro mental; y también incluye acontecimientos encuadrados en el epígrafe de trastornos psiquiátricos: trastorno por déficit de atención/hiperactividad, estado confusional, delirio, desorientación, trastorno de la lectura). Dentro de estos efectos, los términos encuadrados en trastornos del sistema nervioso se notificaron con más frecuencia que los términos encuadrados en el epígrafe de trastornos psiquiátricos.
f Neuropatía periférica (incluye sensación de ardor, disestesia, hormigueo, alteración de la marcha, hipoestesia, disfunción motora, debilidad muscular, neuralgia, neuropatía periférica, neurotoxicidad, parestesia, neuropatía motora periférica, neuropatía sensorial periférica, parálisis del nervio peroneo, alteración sensorial).
g Efectos sobre el habla (disartria, bradilalia, trastorno del habla).
h Trastorno de la visión (incluye diplopía, fotofobia, fotopsia, visión borrosa, disminución de la agudeza visual, deficiencia visual, moscas volantes).
i Neumonitis (incluye enfermedad pulmonar intersticial, opacidad pulmonar, neumonitis).
j Erupción (incluye dermatitis acneiforme, erupción maculopapular, erupción pruriginosa, erupción).
k Mialgia (incluye dolor musculoesquelético, mialgia).
l Edema (incluye edema generalizado, edema, edema periférico, hinchazón periférica, hinchazón).
m Cansancio (incluye astenia, cansancio).
Descripción de las reacciones adversas seleccionadas
Hipercolesterolemia/hipertrigliceridemia
Las reacciones adversas de aumento de los niveles de colesterol o triglicéridos séricos se notificaron en el 81,1% y el 67,2% de los pacientes, respectivamente. De ellos, se produjeron reacciones adversas leves o moderadas de hipercolesterolemia o hipertrigliceridemia en el 62,8% y el 47,9% de los pacientes, respectivamente (ver sección 4.4). La mediana del tiempo hasta el inicio de la hipercolesterolemia y la hipertrigliceridemia fue de 15 días (hipercolesterolemia rango de 1 a 784 días; hipertrigliceridemia rango de 1 a 796 días). La mediana de la duración de la hipercolesterolemia y la hipertrigliceridemia fue de 451 y 427 días, respectivamente.
Efectos sobre el sistema nervioso central
Las reacciones adversas del SNC fueron principalmente efectos cognitivos (27,7%), efectos sobre el estado de ánimo (21,0%), efectos sobre el habla (8,2%) y efectos psicóticos (6,5%), y fueron generalmente leves, transitorios y reversibles espontáneamente al retrasar la dosis y/o reducir la dosis (ver las secciones 4.2 y 4.4). El efecto cognitivo de cualquier grado más frecuente fue el deterioro de la memoria (11,3%), y las reacciones de grado 3 o 4 más frecuentes fueron el estado confusional y el trastorno cognitivo (1,7% y 0,8%, respectivamente). El efecto sobre el estado de ánimo de cualquier grado más frecuente fue la ansiedad (6,5%), y las reacciones de grado 3 y 4 más frecuentes fueron la irritabilidad y la depresión (0,8% y 0,4%, respectivamente). El efecto sobre el habla de cualquier grado más frecuente fue la disartria (4,0%), y las reacciones de grado 3 o 4 fueron la disartria, la bradilalia y el trastorno del habla (0,2% en cada caso). El efecto psicótico de cualquier grado más frecuente fueron alucinaciones (3,7%) y las reacciones de grado 3 o 4 más frecuentes fueron alucinaciones, alucinaciones auditivas y alucinaciones visuales (0,3% en cada caso). La mediana del tiempo hasta el inicio de los efectos cognitivos, los efectos sobre el estado de ánimo, el habla y los efectos psicóticos fue de 109, 43, 49 y 23 días, respectivamente. La mediana de la duración de los efectos cognitivos, del estado de ánimo, del habla y de los efectos psicóticos fue de 223, 143, 147 y 74 días, respectivamente.
Hipertensión arterial
Se notificaron reacciones adversas de hipertensión arterial en el 13% de los pacientes del estudio A y del estudio CROWN (B7461006). De ellos, se produjeron reacciones adversas leves o moderadas de hipertensión arterial en el 6,9% de los pacientes (ver sección 4.4). La mediana del tiempo hasta el inicio de la hipertensión arterial fue de 208 días (rango de 1 a 1 028 días). La mediana de la duración de la hipertensión arterial fue de 219 días.
Hiperglucemia
Se notificaron reacciones adversas de hiperglucemia en el 9,2% de los pacientes del estudio A y del estudio CROWN (B7461006). De ellos, se produjeron reacciones adversas leves o moderadas de hiperglucemia en el 6,1% de los pacientes (ver sección 4.4). La mediana del tiempo hasta el inicio de la hiperglucemia fue de 145 días (rango de 1 a 1 058 días). La mediana de la duración de la hiperglucemia fue de 113 días.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es.
4.9 - Sobredosificación de LORVIQUA 100 mg Comp. recub. con película
El tratamiento de la sobredosis con el medicamento consiste en medidas generales de apoyo. Dado el efecto dependiente de la dosis en el intervalo PR, se recomienda monitorizar el ECG. No existe ningún antídoto para lorlatinib.
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de LORVIQUA 100 mg Comp. recub. con película
Grupo farmacoterapéutico: agentes antineoplásicos, inhibidores de la proteína quinasa, código ATC: L01ED05
Mecanismo de acción
Lorlatinib es un inhibidor competitivo de la adenosina trifosfato (ATP) selectivo de las tirosinas quinasas ALK y oncogén c-ros 1 (ROS1).
En los estudios no clínicos, lorlatinib fue un inhibidor de las actividades catalíticas de la ALK no mutada y de quinasas mutantes de la ALK clínicamente relevantes en análisis con enzimas recombinantes y en células aisladas. Lorlatinib demostró una actividad antitumoral notable en ratones portadores de xenoinjertos tumorales que expresaban fusiones de la proteína 4 asociada al microtúbulo de equinodermo (EML4) con la variante 1 de la ALK (v1), incluidas las mutaciones de la ALK L1196M, G1269A, G1202R e I1171T. Se sabe que dos de estos mutantes de la ALK, G1202R e I1171T, confieren resistencia a alectinib, brigatinib, ceritinib y crizotinib. Lorlatinib también fue capaz de atravesar la barrera hematoencefálica. Lorlatinib demostró actividad en ratones con implantes ortotópicos de tumores cerebrales EML4-ALK o EML4-ALKL1196M.
Eficacia clínica
CPNM avanzado positivo para ALK sin tratamiento previo (estudio CROWN)
La eficacia de lorlatinib para el tratamiento de pacientes con CPNM positivo para la ALK que no habían recibido tratamiento sistémico previo para la enfermedad metastásica se estableció en el estudio B7461006 multicéntrico, abierto, aleatorizado y controlado con tratamiento activo (estudio CROWN). Se requería que los pacientes tuvieran un estado funcional del Grupo Oncológico Cooperativo de la Costa Este (ECOG, por sus siglas en inglés) de 0-2 y CPNM positivo para ALK según lo identificado mediante la técnica VENTANA ALK (D5F3) CDx. Fueron elegibles los pacientes neurológicamente estables con metástasis en el SNC asintomáticas tratadas o no tratadas, incluidas las metástasis leptomeníngeas. Los pacientes debían haber finalizado la radioterapia, incluida la irradiación cerebral parcial o estereotáctica, en las 2 semanas anteriores a la aleatorización; irradiación de todo el cerebro dentro de las 4 semanas previas a la aleatorización.
Los pacientes fueron aleatorizados en una proporción 1:1 para recibir lorlatinib 100 mg por vía oral una vez al día o crizotinib 250 mg por vía oral dos veces al día. La aleatorización se estratificó por origen étnico (asiático frente a no asiático) y por la presencia o ausencia de metástasis en el SNC al inicio del estudio. El tratamiento en ambos grupos continuó hasta la progresión de la enfermedad o hasta que se presentó una toxicidad inaceptable. La variable primaria de eficacia fue la supervivencia libre de progresión (PFS, por sus siglas en inglés) según lo determinado por la revisión central independiente y ciega (BICR, por sus siglas en inglés) de acuerdo a los criterios de evaluación de respuesta en tumores sólidos (RECIST, por sus siglas en inglés) versión 1.1 (v1.1). Otras variables de eficacia fueron la supervivencia global (OS, por sus siglas en inglés), la PFS según la evaluación del investigador, la supervivencia libre de progresión tras el segundo tratamiento (PFS2) y los datos relacionados con la evaluación del tumor por la BICR, incluida la tasa de respuesta objetiva (ORR, por sus siglas en inglés), la duración de la respuesta (DOR, por sus siglas en inglés) y el tiempo hasta la progresión intracraneal (IC-TTP, por sus siglas en inglés). En pacientes con metástasis en el SNC al inicio del estudio, las variables de eficacia adicionales fueron la tasa de respuesta objetiva intracraneal (IC-ORR, por sus siglas en inglés) y la duración de la respuesta intracraneal (IC-DOR, por sus siglas en inglés), todas mediante la BICR.
Un total de 296 pacientes fueron aleatorizados a lorlatinib (n = 149) o a crizotinib (n = 147). Las características demográficas de la población total del estudio fueron: mediana de edad de 59 años (rango de 26 a 90 años), edad ≥ 65 años (35%), 59% mujeres, 49% blancos, 44% asiáticos y 0,3% negros. La mayoría de los pacientes tenían adenocarcinoma (95%) y nunca habían fumado (59%). Las metástasis del sistema nervioso central determinadas por los neurorradiólogos de la BICR estaban presentes en el 26% (n = 78) de los pacientes; de estos, 30 pacientes tenían lesiones medibles en el SNC.
Los resultados del estudio CROWN se resumen en la tabla 3. En el momento del punto de corte de datos, los datos de OS y PFS2 no estaban maduros.
Tabla 3. Resultados globales de eficacia en el estudio CROWN
| Parámetros de eficacia | Lorlatinib N = 149 | Crizotinib N = 147 |
| Duración media del seguimiento, meses (IC del 95%)a | 18 (16, 20) | 15 (13, 18) |
| Supervivencia libre de progresión mediante la BICR | ||
| Número de pacientes con acontecimientos, n (%) | 41 (28%) | 86 (59%) |
| Enfermedad progresiva, n (%) | 32 (22%) | 82 (56%) |
| Muerte, n (%) | 9 (6%) | 4 (3%) |
| Mediana, meses (IC del 95%)a | NE (NE, NE) | 9 (8, 11) |
| Cociente de riesgos (Hazard ratio) (IC del 95%)b | 0,28 (0,19; 0,41) | |
| Valor p* | <0,0001 | |
| Supervivencia global | ||
| Número de pacientes con acontecimientos, n (%) | 23 (15%) | 28 (19%) |
| Mediana, meses (IC del 95%)a | NE (NE, NE) | NE (NE, NE) |
| Cociente de riesgos (Hazard ratio) (IC del 95%)b | 0,72 (0,41; 1,25) | |
| Supervivencia libre de progresión mediante la EI | ||
| Número de pacientes con acontecimientos, n (%) | 40 (27%) | 104 (71%) |
| Enfermedad progresiva, n (%) | 34 (23%) | 99 (67%) |
| Muerte, n (%) | 6 (4%) | 5 (3%) |
| Mediana, meses (IC del 95%)a | NE (NE, NE) | 9 (7, 11) |
| Cociente de riesgos (Hazard ratio) (IC del 95%)b | 0,21 (0,14; 0,31) | |
| Valor p* | <0,0001 | |
| Respuesta global mediante la BICR | ||
| Tasa de respuesta global, n (%) | 113 (76%) | 85 (58%) |
| (IC del 95%)c | (68, 83) | (49, 66) |
| Tiempo hasta la progresión intracraneal | ||
| Mediana, meses (IC del 95%)a | NE (NE, NE) | 16,6 (11, NE) |
| Cociente de riesgos (Hazard ratio) (IC del 95%)b | 0,07 (0,03; 0,17) | |
| Duración de la respuesta | ||
| Número de pacientes que respondieron | 113 | 85 |
| Mediana, meses (IC del 95%)a | NE (NE, NE) | 11 (9, 13) |
| Respuesta global intracraneal en pacientes con lesiones medibles en el SNC al inicio del estudio | N = 17 | N = 13 |
| Tasa de respuesta intracraneal, n (%) | 14 (82%) | 3 (23%) |
| (IC del 95%)c | (57, 96) | (5, 54) |
| Tasa de respuesta global | 71% | 8% |
| Duración de la respuesta | ||
| Número de pacientes que respondieron | 14 | 3 |
| Mediana, meses (IC del 95%)a | NE (NE, NE) | 10 (9, 11) |
| Respuesta global intracraneal en pacientes con cualquier lesión medible o no medible en el SNC al inicio del estudio | N = 38 | N = 40 |
| Tasa de respuesta intracraneal, n (%) | 25 (66%) | 8 (20%) |
| (IC del 95%)c | (49, 80) | (9, 36) |
| Tasa de respuesta global | 61% | 15% |
| Duración de la respuesta | ||
| Número de pacientes que respondieron | 25 | 8 |
| Mediana, meses (IC del 95%)a | NE (NE, NE) | 9 (6, 11) |
Abreviaturas: BICR = revisión central independiente y ciega; IC = intervalo de confianza; SNC = sistema nervioso central; EI = evaluación del investigador; N/n = número de pacientes; NE = no estimable.
* Valor p según la prueba del orden logarítmico estratificada unilateral.
a Basado en el método Brookmeyer-Crowley.
b Cociente de riesgos (Hazard ratio) basado en el modelo de riesgos proporcionales de Cox; bajo riesgos proporcionales, un cociente de riesgos < 1 indica una reducción en la tasa de riesgos a favor de lorlatinib.
c Utilizando el método exacto según la distribución binomial.
Figura 1. Gráfico de Kaplan-Meier de supervivencia libre de progresión mediante la revisión
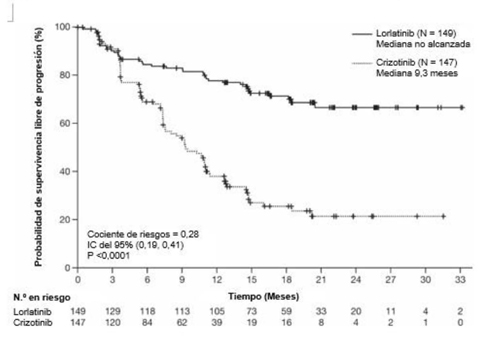
Abreviaturas: IC = intervalo de confianza; N/n = número de pacientes.
El beneficio del tratamiento con lorlatinib fue comparable entre los subgrupos de pacientes y las características de la enfermedad al inicio del estudio, incluidos los pacientes con metástasis en el SNC al inicio del estudio (n = 38, CR = 0,2, IC del 95%: 0,10-0,43) y los pacientes sin metástasis en el SNC al inicio del estudio (n = 111, CR = 0,32, IC del 95%: 0,20-0,49).
CPNM avanzado positivo para ALK tratado previamente con un inhibidor de la quinasa ALK
El uso de lorlatinib en el tratamiento del CPNM avanzado ALK positivo tras el tratamiento con al menos un TKI ALK de segunda generación se investigó en el estudio A, un estudio en fase 1/2 multicéntrico de un solo grupo. Se incluyeron un total de 139 pacientes con CPNM avanzado ALK positivo tras el tratamiento con al menos un TKI ALK de segunda generación en la fase 2 del estudio. Los pacientes recibieron lorlatinib por vía oral a la dosis recomendada de 100 mg una vez al día de manera continua.
La variable primaria de eficacia en la fase 2 del estudio fue la ORR, incluida la intracraneal (IC)-ORR, según la revisión central independiente (ICR, por sus siglas en inglés) y de acuerdo a los RECIST v 1.1 modificados. Las variables secundarias incluyeron la DOR, IC-DOR, el tiempo hasta la respuesta tumoral (TTR, por sus siglas en inglés) y la PFS.
Los datos demográficos de los 139 pacientes con CPNM avanzado ALK positivo tras el tratamiento con al menos un TKI ALK de segunda generación fueron un 56% mujeres, un 48% blancos, un 38% asiáticos y la edad media fue de 53 años (rango: de 29 a 83 años) con un 16% de los pacientes ≥65años de edad. El estado funcional del ECOG al inicio del estudio fue 0 o 1 en el 96% de los pacientes. Las metástasis cerebrales estaban presentes al inicio del estudio en el 67% de los pacientes. De los 139 pacientes, el 20% recibió un TKI ALK previo, excepto crizotinib, el 47% recibió dos TKI ALK previos y el 33% recibió tres o más TKI ALK previos.
Los principales resultados de eficacia del estudio A se incluyen en las tablas 4 y 5.
Tabla 4. Resultados de eficacia global en el estudio A según el tratamiento previo
| Variable de eficacia | Un TKI ALKa previo con o sin quimioterapia previa (n = 28) | Dos o más TKI ALK previos con o sin quimioterapia previa (n = 111) |
| Respuesta parcial, n | 11 | 42 |
| (IC del 95%) | (4,2 - NA) | (5,7 - 24,4) |
| (IC del 95%) | (2,9 - 8,2) | (5,4 - 9,5) |
Abreviaturas: ALK = quinasa del linfoma anaplásico; IC = intervalo de confianza; ICR = revisión central independiente; N/n = número de pacientes; NA = no alcanzado; TKI = inhibidor de tirosina quinasa.
a Alectinib, brigatinib o ceritinib.
b Según la ICR.
Tabla 5. Resultados de eficacia intracraneal* en el estudio A según el tratamiento previo
| Variable de eficacia | Un TKI ALKa previo con o sin quimioterapia previa (n = 9) | Dos o más TKI ALK previos con o sin quimioterapia previa (n = 48) |
| Respuesta parcial, n | 4 | 15 |
| Mediana, meses (IC del 95%) | NA (4,1 - NA) | 12,4 (6,0 - NA) |
Abreviaturas: ALK = quinasa del linfoma anaplásico; IC = intervalo de confianza; ICR = revisión central independiente; N/n = número de pacientes; NA = no alcanzado; TKI = inhibidor de tirosina quinasa.
* En pacientes con al menos una metástasis cerebral cuantificable al inicio del estudio.
a Alectinib, brigatinib o ceritinib.
b Según la ICR.
En la población de eficacia global de 139 pacientes, 56 pacientes tuvieron una respuesta objetiva confirmada por la ICR y una mediana de la TTR de 1,4 meses (rango: de 1,2 a 16,6 meses). La ORR para los asiáticos fue del 49,1% (IC del 95%: 35,1 - 63,2) y del 31,5% para los no asiáticos (IC del 95%: 21,1 - 43,4). Entre los 31 pacientes con una respuesta tumoral IC y al menos una metástasis cerebral cuantificable al inicio del estudio confirmadas por ICR, la media de la IC-TTR fue de 1,4 meses (rango: de 1,2 a 16,2 meses). El IC de la ORR fue del 54,5% para los asiáticos (IC del 95%: 32,2 - 75,6) y del 46,4% para los no asiáticos (IC del 95%: 27,5 - 66,1).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con lorlatinib en todos los grupos de la población pediátrica en cáncer de pulmón (cáncer de células pequeñas y cáncer no microcítico) (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
Este medicamento se ha autorizado con una «aprobación condicional». Esta modalidad de aprobación significa que se espera obtener más información sobre este medicamento.
La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año y esta ficha técnica o resumen de las características del producto (RCP) se actualizará cuando sea necesario.
5.2 - Propiedades farmacocinéticas de LORVIQUA 100 mg Comp. recub. con película
Absorción
Las concentraciones plasmáticas máximas de lorlatinib se alcanzan rápidamente con una mediana de Tmáx de 1,2 horas tras una dosis única de 100 mg y de 2,0 horas tras la administración múltiple de 100 mg una vez al día.
Tras la administración oral de comprimidos de lorlatinib, la biodisponibilidad absoluta media es del 80,8% (IC del 90%: 75,7 - 86,2) en comparación con la administración intravenosa.
La administración de lorlatinib con una comida alta en grasas y alta en calorías dio como resultado una exposición un 5% mayor en comparación con la administración en ayunas. Lorlatinib se puede administrar con o sin alimentos.
A una dosis de 100 mg una vez al día, la media geométrica de la concentración plasmática máxima (% del coeficiente de variación [CV]) fue de 577 (42) ng/ml y el AUC24 fue de 5 650 (39) ng h/ml en pacientes con cáncer. La media geométrica (% CV) del aclaramiento oral fue de 17,7 (39) l/h.
Distribución
La unión in vitro de lorlatinib a las proteínas plasmáticas humanas es del 66% con una unión moderada a la albúmina o a la α1-glicoproteína ácida.
Biotransformación
En los seres humanos, las principales vías metabólicas de lorlatinib son la oxidación y la glucuronidación. Los datos in vitro indican que lorlatinib se metaboliza principalmente por el CYP3A4 y la UGT1A4, con una contribución menor del CYP2C8, CYP2C19, CYP3A5 y la UGT1A3.
En plasma, se observó un metabolito de ácido benzoico de lorlatinib resultante de la escisión oxidativa de los enlaces amida y éter aromático de lorlatinib como un metabolito principal, que representa el 21% de la radioactividad circulante. El metabolito resultante de la escisión oxidativa es farmacológicamente inactivo.
Eliminación
La semivida plasmática de lorlatinib después de una dosis única de 100 mg fue de 23,6 horas. La semivida plasmática efectiva estimada de lorlatinib en el estado estacionario tras la finalización de la autoinducción fue de 14,83 horas. Tras la administración oral de una dosis de 100 mg de lorlatinib radiomarcada, se recuperó una media del 47,7% de la radioactividad en orina y el 40,9% de la radiactividad se recuperó en heces, con una recuperación media total global del 88,6%.
Lorlatinib inalterado fue el principal componente en el plasma y las heces en seres humanos, lo que representa el 44% y el 9,1% de la radiactividad total, respectivamente. Menos del 1% de lorlatinib inalterado se detectó en la orina.
Además, lorlatinib es un inductor a través del receptor X de pregnano (PXR) humano y el receptor constitutivo de androstano (CAR) humano.
Linealidad/No linealidad
A una dosis única, la exposición sistémica a lorlatinib (AUCinf y Cmáx) aumentó en función de la dosis en un rango de dosis de 10 a 200 mg. Se dispone de pocos datos en el rango de dosis de 10 a 200 mg; sin embargo, no se observó desviación de la linealidad en el AUCinf y la Cmáx tras una dosis única.
Tras la administración de dosis múltiples una vez al día, la Cmáx de lorlatinib aumentó proporcionalmente a la dosis mientras que el AUCΤ aumentó de una forma casi proporcional (ligeramente menor) en el rango de dosis de 10 a 200 mg una vez al día.
Además, las exposiciones plasmáticas de lorlatinib en el estado estacionario son inferiores a las esperadas a partir de la farmacocinética de dosis única, lo que indica un efecto de autoinducción dependiente del tiempo neto.
Insuficiencia hepática
Dado que lorlatinib se metaboliza en el hígado, es probable que la insuficiencia hepática aumente las concentraciones plasmáticas de lorlatinib. Los estudios clínicos que se llevaron a cabo excluyeron a pacientes con AST o ALT >2,5 × LSN, o si padecían una neoplasia maligna subyacente, >5,0 × LSN o con bilirrubina total >1,5 × LSN. Los análisis farmacocinéticos poblacionales han demostrado que la exposición a lorlatinib no se alteró clínicamente de manera significativa en pacientes con insuficiencia hepática leve (n = 50). No se recomienda ajustar la dosis en pacientes con insuficiencia hepática leve. No se dispone de información en pacientes con insuficiencia hepática moderada o grave.
Insuficiencia renal
Menos del 1% de la dosis administrada se detecta como lorlatinib inalterado en la orina. Los análisis farmacocinéticos poblacionales han demostrado que la exposición plasmática a lorlatinib en el estado estacionario y los valores de la Cmáx aumentan ligeramente con el empeoramiento de la función renal inicial. Según los resultados de un estudio en pacientes con insuficiencia renal, no se recomienda ajustar la dosis inicial en pacientes con insuficiencia renal leve o moderada [TFGe ajustado a variables de la ecuación de modificación de la dieta en la enfermedad renal (MDRD, por sus siglas en inglés) donde la TFGe (en ml/min/1,73 m2) × área de superficie corporal medida/1,73 ≥30 ml/min]. En este estudio, el AUCinf de lorlatinib aumentó en un 41% en sujetos con insuficiencia renal grave (TFGe absoluta <30 ml/min) en comparación con sujetos con función renal normal (TFGe absoluta ≥90 ml/min). Se recomienda una dosis reducida de lorlatinib en pacientes con insuficiencia renal grave, por ejemplo, una dosis inicial de 75 mg una vez al día por vía oral (ver sección 4.2). No se dispone de información para pacientes en diálisis renal.
Edad, sexo, raza, peso corporal y fenotipo
Los análisis farmacocinéticos poblacionales en pacientes con CPNM avanzado y voluntarios sanos indican que no existen efectos clínicamente relevantes de la edad, el sexo, la raza, el peso corporal y los fenotipos para el CYP3A5 y el CYP2C19.
Electrofisiología cardíaca
En el estudio A, 2 pacientes (0,7%) tuvieron valores de QTc con la corrección de Fridericia (QTcF) absolutos >500 ms, y 5 pacientes (1,8%) tuvieron un cambio en el QTcF desde el inicio del estudio >60 ms.
Además, se evaluó el efecto de una dosis oral única de lorlatinib (50 mg, 75 mg y 100 mg) con y sin 200 mg de itraconazol una vez al día en un estudio cruzado de 2 direcciones en 16 voluntarios sanos. No se observaron incrementos en la media del QTc a las concentraciones medias observadas de lorlatinib en este estudio.
En 295 pacientes que recibieron lorlatinib a la dosis recomendada de 100 mg una vez al día y a los que se les realizó un ECG en el estudio A, el estudio con lorlatinib excluyó a aquellos con un intervalo QTc > 470 ms. En la población de estudio, el cambio medio máximo desde el inicio del estudio para el intervalo PR fue de 16,4 ms (límite superior del IC bilateral del 90% 19,4 ms) (ver las secciones 4.2, 4.4 y 4.8). De ellos, 7 pacientes tenían un intervalo PR inicial >200 ms. Entre los 284 pacientes con intervalo PR <200 ms, el 14% tuvo una prolongación del intervalo PR ≥200 ms después de comenzar el tratamiento con lorlatinib. La prolongación del intervalo PR se produjo de una manera dependiente de la concentración. Se produjo bloqueo auriculoventricular en el 1,0% de los pacientes.
Para aquellos pacientes que presentan prolongación del intervalo PR, puede ser necesaria una modificación de la dosis (ver sección 4.2).
5.3 - Datos preclínicos sobre seguridad de LORVIQUA 100 mg Comp. recub. con película
Toxicidad a dosis repetidas
Las principales toxicidades observadas fueron inflamación en múltiples tejidos (piel y cuello uterino de ratas y pulmón, tráquea, piel, ganglios linfáticos y/o la cavidad oral incluyendo el hueso mandibular de perros, relacionada con aumentos en los glóbulos blancos, fibrinógeno y/o globulina y disminuciones en la albúmina) y cambios en el páncreas (con aumentos de amilasa y lipasa), sistema hepatobiliar (con aumentos de enzimas hepáticas), sistema reproductivo masculino, sistema cardiovascular, riñones y tracto gastrointestinal, nervios periféricos y sistema nervioso central (con potencial para deterioro cognitivo funcional) a una dosis equivalente a la exposición clínica en humanos a la posología recomendada. También se observaron cambios en la tensión arterial y la frecuencia cardíaca, y el complejo QRS y el intervalo PR en animales después de una dosis alta (aproximadamente 2,6 veces la exposición clínica en humanos a 100 mg tras una dosis única según la Cmáx). Todos los hallazgos en el órgano diana con la excepción de la hiperplasia del conducto biliar hepático fueron parcial o totalmente reversibles.
Genotoxicidad
Lorlatinib no es mutagénico pero es aneugénico in vitro e in vivo sin efecto observado para la aneugenicidad a aproximadamente 16,5 veces la exposición clínica en humanos a 100 mg según el AUC.
Carcinogenicidad
No se han realizado estudios de carcinogenicidad con lorlatinib.
Toxicidad para la reproducción
Se observó degeneración de los túbulos seminíferos y/o atrofia en los testículos, así como cambios en el epidídimo (inflamación y/o vacuolación) en ratas y perros. En la próstata, se observó atrofia glandular de mínima a leve en perros a una dosis equivalente a la exposición clínica en humanos a la posología recomendada. Los efectos sobre los órganos reproductivos masculinos fueron parcial o totalmente reversibles.
En los estudios de toxicidad embriofetal realizados en ratas y conejos, respectivamente, se observó aumento de la embrioletalidad así como bajo peso fetal y malformaciones fetales. Las anomalías morfológicas fetales incluyeron miembros rotados, dedos supernumerarios, gastrosquisis, riñones malformados, cabeza abombada, paladar ojival y dilatación de los ventrículos cerebrales. La exposición a las dosis más bajas con efectos embriofetales en animales fue equivalente a la exposición clínica en humanos a 100 mg, según el AUC.
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de LORVIQUA 100 mg Comp. recub. con película
Núcleo del comprimido
Celulosa microcristalina
Hidrogenofosfato de calcio
Almidón glicolato sódico
Estearato de magnesio
Recubrimiento con película
Hipromelosa
Lactosa monohidrato
Macrogol
Triacetina
Dióxido de titanio (E171)
Óxido de hierro negro (E172)
Óxido de hierro rojo (E172)
6.2 - Incompatibilidades de LORVIQUA 100 mg Comp. recub. con película
No procede.
6.3 - Período de validez de LORVIQUA 100 mg Comp. recub. con película
3 años.
6.4 - Precauciones especiales de conservación de LORVIQUA 100 mg Comp. recub. con película
No requiere condiciones especiales de conservación.
6.5 - Naturaleza y contenido del recipiente de LORVIQUA 100 mg Comp. recub. con película
Blísteres de OPA/Al/PVC reforzados con lámina de aluminio con 10 comprimidos recubiertos con película.
Lorviqua 25 mg comprimidos recubiertos con película
Cada envase contiene 90 comprimidos recubiertos con película en 9 blísteres.
Lorviqua 100 mg comprimidos recubiertos con película
Cada envase contiene 30 comprimidos recubiertos con película en 3 blísteres.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de LORVIQUA 100 mg Comp. recub. con película
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Bruxelles
Bélgica
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/19/1355/002
EU/1/19/1355/003
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 06/mayo/2019
Fecha de la última renovación: 04/abril/2022
10. - FECHA DE LA REVISIÓN DEL TEXTO
04/2023
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
PRESENTACIONES Y PRECIO
LORVIQUA 25 mg comprimidos recubiertos con película, 90 comprimidos: PVL: 5.230,00 €; PVP: 5.285,91€; PVP IVA: 5.497,35 €
LORVIQUA 100 mg comprimidos recubiertos con película, 30 comprimidos: PVL: 5.230,00 €; PVP: 5.285,91 €; PVP IVA: 5.497,35 €
CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN
Con receta médica. Diagnóstico hospitalario. Fármaco de Dispensación Hospitalaria sin Cupón Precinto (DIHSC): se limita su dispensación, sin necesidad de visado, a los pacientes no hospitalizados en los Servicios de Farmacia o centros sanitarios autorizados del SNS; por lo tanto irá desprovisto de cupón precinto.
CONDICIONES DE LA PRESTACIÓN FARMACÉUTICA
Financiado por el Sistema Nacional de Salud, sin aportación.
Para información adicional, por favor, contacte con el Centro de Información Médico-Farmacéutica llamando al + 34 914909900 o consulte nuestra página web www.pfizer.es.

