 VANFLYTA 17,7 MG COMPRIMIDOS RECUBIERTOS CON PELICULA
VANFLYTA 17,7 MG COMPRIMIDOS RECUBIERTOS CON PELICULA
| ATC: Quizartinib |
| PA: Quizartinib dihidrocloruro |
Envases
- Env. con 28
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica- Comercializado: No
- Situación: Alta
- Código Nacional: 764565
- EAN13: 8470007645658
- Conservar en frío: No
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
VANFLYTA 17,7 mg Comp. recub. con película
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
VANFLYTA 17,7 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene 17,7 mg de quizartinib (como dihidrocloruro).
VANFLYTA 26,5 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene 26,5 mg de quizartinib (como dihidrocloruro). Para consultar la lista completa de excipientes, ver sección 6.1.
3. - FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido) VANFLYTA 17,7 mg comprimidos recubiertos con película
Comprimidos recubiertos con película de color blanco, redondos, de 8,9 mm de diámetro y con la inscripción “DSC 511” en una cara.
VANFLYTA 26,5 mg comprimidos recubiertos con película
Comprimidos recubiertos con película de color amarillo, redondos, de 10,2 mm de diámetro y con la inscripción “DSC 512” en una cara.
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de VANFLYTA 17,7 mg Comp. recub. con película
VANFLYTA está indicado, en combinación con quimioterapia estándar de inducción con citarabina y antraciclina y con quimioterapia estándar de consolidación con citarabina, seguido de terapia de mantenimiento con VANFLYTA en monoterapia, para pacientes adultos con leucemia mieloide aguda (LMA) de nuevo diagnóstico que sea FLT3-ITD positiva.
4.2 - Posología y administración de VANFLYTA 17,7 mg Comp. recub. con película
4.3 - Contraindicaciones de VANFLYTA 17,7 mg Comp. recub. con película
• Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
• Síndrome de QT largo congénito (ver sección 4.4).
• Lactancia (ver sección 4.6).
4.4 - Advertencias y Precauciones de VANFLYTA 17,7 mg Comp. recub. con película
Prolongación del intervalo QT
Quizartinib se asocia a prolongación del intervalo QT (ver sección 4.8). La prolongación del intervalo QT puede aumentar el riesgo de arritmias ventriculares o de torsade de pointes. Los pacientes con síndrome de QT largo congénito y/o antecedentes de torsade de pointes fueron excluidos del programa de desarrollo de quizartinib. No se debe utilizar VANFLYTA en pacientes con síndrome de QT largo congénito.
VANFLYTA se debe utilizar con precaución en pacientes que corren un riesgo significativo de desarrollar prolongación del intervalo QT. Esto incluye a los pacientes con enfermedad cardiovascular no controlada o importante (p. ej., antecedentes de bloqueo cardiaco de segundo o tercer grado [sin marcapasos], infarto de miocardio en los 6 meses anteriores, angina de pecho no controlada, hipertensión no controlada, insuficiencia cardiaca congestiva, antecedentes de arritmias ventriculares clínicamente relevantes o torsade de pointes) y a los pacientes que reciben medicamentos concomitantes que se sabe que prolongan el intervalo QT. Los electrolitos deben estar dentro de los valores de referencia (ver sección 4.2).
No se debe comenzar el tratamiento con VANFLYTA si el intervalo QTcF es superior a 450 ms. Durante la inducción y la consolidación, se deben realizar ECG antes de iniciar el tratamiento y luego
una vez por semana durante el tratamiento con quizartinib, o con mayor frecuencia si está clínicamente indicado.
Durante el mantenimiento, se deben realizar ECG antes de iniciar el tratamiento y luego una vez por semana durante el primer mes tras el inicio y el aumento de la dosis, y posteriormente si está clínicamente indicado. La dosis inicial de mantenimiento no se debe aumentar si el intervalo QTcF es superior a 450 ms (ver la Tabla 1).
Se debe suspender VANFLYTA de forma permanente en pacientes que desarrollan prolongación del intervalo QT con signos o síntomas de arritmia potencialmente mortal (ver sección 4.2).
El control del intervalo QT mediante ECG se debe realizar con mayor frecuencia en pacientes que corren un riesgo significativo de desarrollar prolongación del intervalo QT y torsade de pointes.
Se debe controlar y corregir la hipocalemia y la hipomagnesemia antes y durante el tratamiento con VANFLYTA. Se deben controlar los electrolitos y realizar ECG con mayor frecuencia en pacientes que presentan diarrea o vómitos.
Control mediante ECG si se utilizan medicamentos que prolongan el intervalo QT
Se debe controlar con mayor frecuencia mediante ECG a los pacientes si se requiere administrar
VANFLYTA junto con medicamentos que se sabe que prolongan el intervalo QT (ver sección 4.5).
Coadministración con inhibidores potentes de CYP3A
Se debe reducir la dosis de VANFLYTA cuando se utiliza de forma concomitante con inhibidores potentes de CYP3A, ya que pueden aumentar la exposición de quizartinib (ver secciones 4.2 y 4.5).
Infecciones en pacientes de edad avanzada
Se han producido infecciones mortales con quizartinib con mayor frecuencia en pacientes de edad avanzada (es decir, mayores de 65 años), en comparación con pacientes más jóvenes, especialmente al principio del periodo de tratamiento. Se debe vigilar estrechamente a los pacientes mayores de 65 años por si aparecen infecciones graves durante la inducción.
Mujeres en edad fértil/anticonceptivos en hombres y mujeres
Según los hallazgos observados en animales, quizartinib puede producir daños embriofetales si se administra a una mujer embarazada. Las mujeres en edad fértil se deben realizar pruebas de embarazo en los 7 días anteriores a comenzar el tratamiento con VANFLYTA. Las mujeres en edad fértil deben utilizar anticonceptivos efectivos durante el tratamiento con VANFLYTA y durante al menos 7 meses después de la última dosis. Los hombres cuyas parejas estén en edad fértil deben utilizar anticonceptivos efectivos durante el tratamiento con VANFLYTA y durante al menos 4 meses después de la última dosis (ver sección 4.6).
Tarjeta de información para el paciente
El médico prescriptor debe comentar los riesgos del tratamiento con VANFLYTA con el paciente. El paciente recibirá la tarjeta de información para el paciente con cada prescripción (incluida en el envase del medicamento).
4.5 - Interacciones con otros medicamentos de VANFLYTA 17,7 mg Comp. recub. con película
Quizartinib y su metabolito activo AC886 se metabolizan principalmente por CYP3A in vitro. Efecto de otros medicamentos en VANFLYTA
Inhibidores potentes de CYP3A/glucoproteína P (P-gp)
La administración conjunta de ketoconazol (200 mg dos veces al día durante 28 días), un inhibidor potente de CYP3A/P-gp, con una dosis única de VANFLYTA aumentó la concentración plasmática máxima (Cmáx) y el área bajo la curva (AUCinf) de quizartinib en 1,17 veces y 1,94 veces, respectivamente, y disminuyó la Cmáx y el AUCinf de AC886 en 2,5 veces y 1,18 veces, respectivamente, en comparación con VANFLYTA en monoterapia. En estado estacionario, se calculó que la exposición de quizartinib (Cmáx y AUC0-24h) aumentó en 1,86 veces y 1,96 veces, respectivamente, y la exposición de AC886 (Cmáx y AUC0-24h) disminuyó en 1,22 veces y 1,17 veces, respectivamente. El aumento de la exposición de quizartinib puede aumentar el riesgo de toxicidad.
Se debe reducir la dosis de VANFLYTA, como se muestra en la tabla siguiente, si no se puede evitar el uso concomitante con inhibidores potentes de CYP3A. Para obtener más información sobre los ajustes de dosis, ver la Tabla 3 en la sección 4.2.
| Dosis completa | Reducciones de dosis en el uso concomitante con inhibidores potentes de CYP3A |
| 26,5 mg | 17,7 mg |
| 35,4 mg | 17,7 mg |
| 53 mg | 26,5 mg |
Entre los inhibidores potentes de CYP3A/P-gp se encuentran itraconazol, posaconazol, voriconazol, claritromicina, nefazodona, telitromicina y antirretrovirales (ciertos medicamentos utilizados para tratar el VIH pueden aumentar el riesgo de efectos adversos (p. ej., ritonavir) o reducir la eficacia
(p. ej., efavirenz o etravirina) de VANFLYTA).
Inhibidores moderados de CYP3A
La administración conjunta de fluconazol (200 mg dos veces al día durante 28 días), un inhibidor moderado de CYP3A, con una dosis única de VANFLYTA aumentó la Cmáx de quizartinib y AC886 en 1,11 veces y 1,02 veces, respectivamente, y el AUCinf en 1,20 veces y 1,14 veces, respectivamente. Este cambio no se consideró clínicamente relevante. No se recomienda modificar la dosis.
Inductores potentes o moderados de CYP3A
La administración conjunta de efavirenz (tratamiento inicial a 600 mg una vez al día durante 14 días),
un inductor moderado de CYP3A, con una dosis única de VANFLYTA disminuyó la Cmáx y el AUCinf
de quizartinib en aproximadamente 1,18 veces y 9,7 veces, respectivamente, en comparación con
VANFLYTA en monoterapia. La Cmáx y el AUCinf de AC886 disminuyeron en aproximadamente
3,1 veces y 26 veces, respectivamente (ver sección 5.2).
Una reducción de la exposición de quizartinib puede dar lugar a una reducción de la eficacia. Se debe evitar la administración conjunta de VANFLYTA con inductores potentes o moderados de CYP3A.
Entre los inductores potentes de CYP3A4 se encuentran apalutamida, carbamazepina, enzalutamida, mitotano, fenitoína, rifampicina y ciertos medicamentos a base de plantas como la hierba de San Juan (también conocida como Hypericum perforatum). Entre los inductores moderados de CYP3A4 se encuentran efavirenz, bosentán, etravirina, fenobarbital y primidona.
Medicamentos que prolongan el intervalo QT
La administración conjunta de VANFLYTA con otros medicamentos que prolongan el intervalo QT puede aumentar aún más la incidencia de prolongación del intervalo QT. Entre los medicamentos que prolongan el intervalo QT se encuentran los azoles antifúngicos, ondansetrón, granisetrón, azitromicina, pentamidina, doxiciclina, moxifloxacino, atovacuona, proclorperazina y tacrólimus. Se debe tener precaución al administrar medicamentos que prolongan el intervalo QT junto con VANFLYTA (ver sección 4.4).
Medicamentos que reducen los ácidos gástricos
El inhibidor de la bomba de protones lansoprazol disminuyó la Cmáx y el AUCinf de quizartinib en
1,16 veces y 1,05 veces, respectivamente. Este cambio en la absorción de quizartinib no se consideró
clínicamente relevante. No se recomienda modificar la dosis.
Efecto de VANFLYTA en otros medicamentos
Sustratos de la glucoproteína P (P-gp)
La administración conjunta de quizartinib y dabigatrán etexilato (un sustrato de la P-gp) aumentó la
Cmáx total y libre de dabigatrán en 1,12 veces y 1,13 veces, respectivamente, y aumentó el AUCinf total y libre de dabigatrán en 1,13 veces y 1,11 veces, respectivamente (ver sección 5.2). Quizartinib es un
inhibidor débil de la P-gp, y no se recomienda modificar la dosis cuando se administran sustratos de la
P-gp junto con VANFLYTA.
Sustratos de la proteína de resistencia al cáncer de mama (BCRP)
Los datos in vitro indican que quizartinib es un inhibidor de la BCRP. Actualmente, se desconoce la relevancia clínica de este hallazgo. Se debe tener precaución al administrar quizartinib junto con medicamentos que son sustratos de la BCRP.
4.6 - Embarazo y Lactancia de VANFLYTA 17,7 mg Comp. recub. con película
Mujeres en edad fértil/anticonceptivos en hombres y mujeres
Las mujeres en edad fértil se deben realizar pruebas de embarazo en los 7 días anteriores a comenzar el tratamiento con VANFLYTA.
Quizartinib puede producir daños embriofetales cuando se administra a una mujer embarazada (ver sección 5.3). Por lo tanto, las mujeres en edad fértil deben utilizar anticonceptivos efectivos durante el tratamiento con VANFLYTA y durante al menos 7 meses después de la última dosis.
Los hombres cuyas parejas están en edad fértil deben utilizar anticonceptivos efectivos durante el tratamiento con VANFLYTA y durante al menos 4 meses después de la última dosis.
Embarazo
No hay datos relativos al uso de quizartinib en mujeres embarazadas. En función de los hallazgos en animales, quizartinib puede producir toxicidad embriofetal si se administra a mujeres embarazadas (ver sección 5.3).
No debe utilizarse VANFLYTA durante el embarazo ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos, a no ser que la situación clínica de la mujer requiera tratamiento. Se debe informar a las mujeres embarazadas sobre el posible riesgo para el feto.
Lactancia
Se desconoce si quizartinib o sus metabolitos activos se excretan en la leche materna. No se puede excluir el riesgo en niños lactantes. Debido al potencial de reacciones adversas graves en los lactantes, las mujeres no deben dar el pecho durante el tratamiento con VANFLYTA ni durante al menos
5 semanas después de la última dosis (ver sección 4.3).
Fertilidad
No hay datos en seres humanos relativos al efecto de quizartinib en la fertilidad. En función de los hallazgos en animales, el tratamiento con VANFLYTA puede afectar a la fertilidad en mujeres y en hombres (ver sección 5.3).
4.7 - Efectos sobre la capacidad de conducción de VANFLYTA 17,7 mg Comp. recub. con película
La influencia de VANFLYTA sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 - Reacciones Adversas de VANFLYTA 17,7 mg Comp. recub. con película
Resumen del perfil de seguridad
Las reacciones adversas más frecuentes fueron alanina aminotransferasa elevada (58,9 %), recuento plaquetario disminuido (40,0 %), disminución de hemoglobina (37,4 %), diarrea (37,0 %), náuseas (34,0 %), dolor abdominal (29,4 %), cefalea (27,5 %), vómitos (24,5 %) y recuento de neutrófilos disminuido (21,9 %).
Las reacciones adversas más frecuentes de grado 3 o 4 fueron recuento plaquetario disminuido (40 %), disminución de hemoglobina (35,5 %), recuento de neutrófilos disminuido (21,5 %), alanina aminotransferasa elevada (12,1 %), bacteriemia (7,2 %) e infecciones por hongos (5,7 %). Las reacciones adversas graves más frecuentes en el grupo de VANFLYTA fueron neutropenia (3,0 %), infecciones por hongos (2,3 %) e infecciones por herpes (2,3 %). Las reacciones adversas con desenlace mortal fueron infecciones por hongos (0,8 %) y parada cardiaca (0,4 %).
Las reacciones adversas más frecuentes asociadas a la interrupción de la dosis de VANFLYTA fueron neutropenia (10,6 %), trombocitopenia (4,5 %) e intervalo QT largo en el electrocardiograma (2,6 %). Las reacciones adversas más frecuentes asociadas a la reducción de la dosis fueron neutropenia
(9,1 %), trombocitopenia (4,5 %) e intervalo QT largo en el electrocardiograma (3,8 %).
La reacción adversa más frecuente asociada a la suspensión permanente de VANFLYTA fue trombocitopenia (1,1 %).
Tabla de reacciones adversas
Se investigó la seguridad de VANFLYTA en QuANTUM-First, un estudio doble ciego, aleatorizado y controlado con placebo en pacientes adultos con LMA FLT3-ITD positiva de nuevo diagnóstico.
Las reacciones adversas se enumeran de acuerdo con la clasificación por órganos y sistemas de MedDRA. Dentro de cada clasificación por órganos y sistemas, las reacciones adversas se presentan por frecuencia, con las reacciones más frecuentes en primer lugar, utilizando la siguiente convención: muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes (≥1/1000 a <1/100), raras
(≥1/10 000 a <1/1000), muy raras (<1/10 000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada categoría de frecuencia, las reacciones adversas se presentan en
orden descendente de gravedad.
Tabla 4: Reacciones adversas
| Reacción adversa | Todos los grados % | Grado 3 o 4 % | Categoría de frecuencia (todos los grados) |
| Infecciones e infestaciones | |||
| Infección del tracto respiratorio superiora | 18,1 | 1,9 | Muy frecuentes |
| Infecciones por hongosb | 15,1 | 5,7 | Muy frecuentes |
| Infecciones por herpesc | 14,0 | 3,0 | Muy frecuentes |
| Bacteriemiad | 11,3 | 7,2 | Muy frecuentes |
| Trastornos de la sangre y del sistema linfático | |||
| Trombocitopeniae | 40,0 | 40,0 | Muy frecuentes |
| Anemiae | 37,4 | 35,5 | Muy frecuentes |
| Neutropeniae | 21,9 | 21,5 | Muy frecuentes |
| Pancitopenia | 2,6 | 2,3 | Frecuentes |
| Trastornos del metabolismo y de la nutrición | |||
| Apetito disminuido | 17,4 | 4,9 | Muy frecuentes |
| Trastornos del sistema nervioso | |||
| Cefaleaf | 27,5 | 0 | Muy frecuentes |
| Trastornos cardiacos | |||
| Parada cardiacag | 0,8 | 0,4 | Poco frecuentes |
| Fibrilación ventricularg | 0,4 | 0,4 | Poco frecuentes |
| Trastornos respiratorios, torácicos y mediastínicos | |||
| Epistaxis | 15,1 | 1,1 | Muy frecuentes |
| Trastornos gastrointestinales | |||
| Diarreah | 37,0 | 3,8 | Muy frecuentes |
| Náuseas | 34,0 | 1,5 | Muy frecuentes |
| Dolor abdominali | 29,4 | 2,3 | Muy frecuentes |
| Vómitos | 24,5 | 0 | Muy frecuentes |
| Dispepsia | 11,3 | 0,4 | Muy frecuentes |
| Trastornos hepatobiliares | |||
| ALT elevadae | 58,9 | 12,1 | Muy frecuentes |
| Trastornos generales y alteraciones en el lugar de administración | |||
| Edemaj | 18,9 | 0,4 | Muy frecuentes |
| Exploraciones complementarias | |||
| QT largo en el electrocardiogramak | 14,0 | 3,0 | Muy frecuentes |
Quimioterapia estándar = citarabina (arabinósido de citosina) y antraciclina (daunorubicina o idarubicina).
a Las infecciones del tracto respiratorio superior incluyen infección del tracto respiratorio superior,
nasofaringitis, sinusitis, rinitis, amigdalitis, laringofaringitis, faringitis bacteriana, faringoamigdalitis, faringitis vírica y sinusitis aguda.
b Las infecciones por hongos incluyen candidiasis oral, aspergilosis broncopulmonar, infección por hongos, candidiasis vulvovaginal, infección por Aspergillus, infección fúngica del tracto respiratorio inferior, infección fúngica oral, infección por cándida, infección cutánea por hongos, mucormicosis, candidiasis orofaríngea, aspergilosis oral, infección hepática fúngica, candidiasis hepatoesplénica, onicomicosis, fungemia, cándida sistémica y micosis sistémica.
c Las infecciones por herpes incluyen herpes oral, herpes zóster, infecciones por virus herpes, herpes simple,
infección por herpesvirus humano 6, herpes genital y dermatitis herpética.
d La bacteriemia incluye bacteriemia, bacteriemia por Klebsiella, bacteriemia por estafilococos, bacteriemia enterocócica, bacteriemia estreptocócica, bacteriemia relacionada con dispositivos, bacteriemia por Escherichia, bacteriemia por Corynebacterium y bacteriemia por Pseudomonas.
e Términos basados en datos de laboratorio.
f La cefalea incluye cefalea, cefalea de tensión y migraña.
g Un sujeto presentó dos acontecimientos (fibrilación ventricular y parada cardiaca).
h La diarrea incluye diarrea y diarrea hemorrágica.
d El dolor abdominal incluye dolor abdominal, dolor en la zona superior del abdomen, molestia abdominal, dolor en la zona inferior del abdomen y dolor gastrointestinal.
j El edema incluye edema periférico, edema de cara, edema, sobrecarga de líquidos, edema generalizado, edema localizado con hinchazón periférica e hinchazón facial.
k QT largo en el electrocardiograma incluye QT largo en el electrocardiograma e intervalo QT del
electrocardiograma anormal.
Descripción de reacciones adversas seleccionadas
Trastornos cardiacos
Quizartinib prolonga el intervalo QT en el ECG. Se notificaron reacciones adversas que surgieron durante el tratamiento de prolongación del intervalo QT de cualquier grado en el 14,0 % de los pacientes tratados con VANFLYTA, y el 3,0 % de los pacientes presentó reacciones de grado 3 o más graves. La prolongación del QT se asoció a la reducción de la dosis en 10 (3,8 %) pacientes, a la interrupción de la dosis en 7 (2,6 %) pacientes y a la suspensión en 2 (0,8 %) pacientes. Se observó QTcF >500 ms en el 2,3 % de los pacientes en función de la revisión central de los datos de ECG. Dos (0,8 %) pacientes tratados con VANFLYTA presentaron parada cardiaca con registro de fibrilación ventricular, uno de ellos con desenlace mortal, ambos en el contexto de hipocalemia grave. Se deben realizar ECG y controlar y corregir la hipocalemia y la hipomagnesemia antes y durante el tratamiento con VANFLYTA. Para las modificaciones posológicas en pacientes con prolongación del intervalo QT, ver sección 4.2.
Otras poblaciones especiales
Edad avanzada
Se han producido infecciones mortales con mayor frecuencia con quizartinib en pacientes de edad avanzada (es decir, mayores de 65 años de edad), en comparación con pacientes más jóvenes (13 %
frente a 5,7 %), especialmente en el periodo inicial del tratamiento.
Se debe vigilar estrechamente a los pacientes mayores de 65 años de edad por si aparecen infecciones graves durante la inducción.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 - Sobredosificación de VANFLYTA 17,7 mg Comp. recub. con película
No hay ningún antídoto conocido para las sobredosis de VANFLYTA. En caso de sobredosis importante, se deben proporcionar las medidas de apoyo necesarias, con interrupción del tratamiento, evaluación de la hematología y control mediante ECG, así como prestar atención a los electrolitos séricos y a los medicamentos concomitantes que puedan predisponer a los pacientes a la prolongación del intervalo QT y/o a torsade de pointes. Se debe tratar a los pacientes con cuidados sintomáticos y de apoyo (ver las secciones 4.2 y 4.4).
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de VANFLYTA 17,7 mg Comp. recub. con película
Grupo farmacoterapéutico: Agentes antineoplásicos, inhibidores de la proteína quinasa, código ATC: L01EX11
Mecanismo de acción
Quizartinib es un inhibidor del receptor tirosina quinasa FLT3. Quizartinib y su metabolito principal, AC886, se unen de forma competitiva al lugar de unión de adenosina trifosfato (ATP) de FLT3 con gran afinidad. Quizartinib y AC886 inhiben la actividad de quinasa de FLT3, impidiendo la autofosforilación del receptor y, por tanto, inhibiendo la señalización adicional posterior del receptor FLT3 y bloqueando la proliferación celular dependiente de FLT3-ITD.
Efectos farmacodinámicos
Electrofisiología cardiaca
El análisis de la respuesta a la exposición de QuANTUM-First predijo una prolongación de 24,1 ms
del intervalo QTcF dependiente de la concentración (límite superior del intervalo de confianza [IC] del
90 % bilateral: 26,6 ms) con la Cmáx en estado estacionario de quizartinib (53 mg) durante la terapia de mantenimiento.
Eficacia clínica y seguridad
Se evaluó la eficacia y seguridad de quizartinib frente a placebo en un estudio de fase III, aleatorizado, doble ciego y controlado con placebo, QuANTUM-First. En el estudio participaron 539 pacientes adultos de entre 18 y 75 años de edad (el 25 % tenía 65 años de edad o más), a los que se había diagnosticado recientemente una LMA FLT3-ITD positiva, determinada prospectivamente mediante
un análisis del estudio clínico. Se aleatorizó a los pacientes (1:1) para recibir VANFLYTA 35,4 mg una vez al día (n = 268) o placebo (n = 271) durante dos semanas en cada ciclo en combinación con
quimioterapia estándar (inducción seguida de consolidación para los pacientes que respondían),
seguido de terapia de mantenimiento con VANFLYTA en monoterapia (26,5 mg una vez al día durante dos semanas y 53 mg una vez al día a partir de entonces) o placebo durante un máximo de
36 ciclos (28 días/ciclo).
Los pacientes recibieron hasta 2 ciclos de quimioterapia de inducción con daunorubicina los días 1,
2 y 3 o idarubicina los días 1, 2 y 3 y citarabina durante 7 días, seguida de un tratamiento posterior a la remisión que consistió en hasta 4 ciclos de quimioterapia de consolidación y/o TCMH. La quimioterapia de consolidación consistió en citarabina los días 1, 3 y 5. Los pacientes que se sometieron a un TCMH dejaron de recibir el tratamiento del estudio 7 días antes del inicio del
esquema de acondicionamiento. Consultar la ficha técnica o resumen de las características del producto para las recomendaciones posológicas de daunorubicina, idarubicina y citarabina.
Los dos grupos de tratamiento aleatorizado estaban bien equilibrados con respecto a los datos demográficos, las características de la enfermedad y los factores de estratificación basales. De los
539 pacientes, la mediana de edad era de 56 años de edad (intervalo: 20 a 75 años), el 26,1 % de los
pacientes del grupo de quizartinib y el 24 % de los pacientes del grupo de placebo tenían 65 años de edad o más; el 54,5 % eran mujeres y el 45,5 % hombres; el 59,7 % era de raza blanca, el 29,3 % asiáticos, el 1,3 % de raza negra o afroamericanos y el 9,7 % de otras razas. El 84 % de los pacientes tenía un estado funcional basal de ECOG (Grupo Oncológico Cooperativo del Este) de 0 o 1. La mayoría de los pacientes (72,4 %) tenía un estado de riesgo citogenético basal intermedio. La frecuencia de alelos variantes de FLT3-ITD (VAF) fue del 3-25 % en el 35,6 % de los pacientes, superior al 25-50 % en el 52,1 % de los pacientes y superior al 50 % en el 12,1 % de los pacientes.
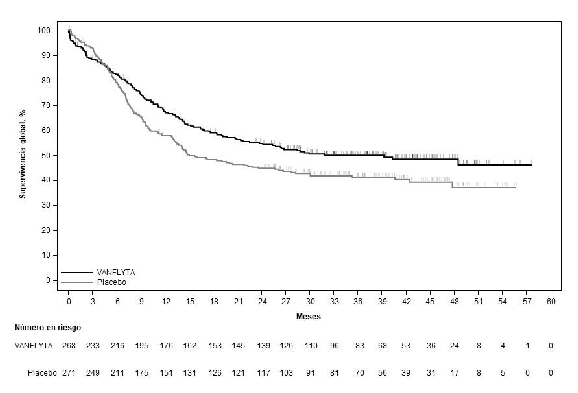
La variable primaria de eficacia fue la supervivencia global (SG) definida como el tiempo desde la aleatorización hasta la muerte por cualquier causa.
El estudio demostró una mejoría estadísticamente significativa en la SG para el grupo de quizartinib
(ver la Tabla 5 y la Figura 1). La mediana de tiempo de seguimiento del estudio fue de 39,2 meses.
Se observó una diferencia entre el grupo de quizartinib frente al grupo de placebo en las estimaciones de las tasas de supervivencia (IC del 95 %) en los momentos de referencia de 12, 24, 36 y 48 meses (ver la Tabla 5).
La tasa de remisión completa (RC) [IC del 95 %] para quizartinib fue del 54,9 % (147/268) [48,7;
60,9] frente al 55,4 % (150/271) [49,2; 61,4] para placebo.
Tabla 5: Resultados de eficacia de QuANTUM-First (población por intención de tratar)
| Quizartinib N = 268 | Placebo N = 271 | |
| SG (meses) | ||
| Mediana (IC del 95 %)a | 31,9 (21,0; NE) | 15,1 (13,2; 26,2) |
| HRb en relación con placebo (IC del 95 %) | 0,776 (0,615; 0,979) | |
| Valor p (prueba de rango logarítmico estratificado bilateral) | 0,0324 | |
| Tasa de SG (%) (IC del 95 %)a | ||
| 12 meses | 67,4 (61,3; 72,7) | 57,7 (51,6; 63,4) |
| 24 meses | 54,7 (48,4; 60,5) | 44,7 (38,7; 50,6) |
| 36 meses | 49,9 (43,7; 55,9) | 41,1 (35,0; 47,0) |
| 48 meses | 48,4 (41,9; 54,5) | 37,0 (29,8; 44,2) |
IC = intervalo de confianza; NE = no estimable
a Estimación de Kaplan-Meier
b El hazard ratio (HR) se basó en el modelo de regresión de Cox estratificado.
Figura 1: Curvas de Kaplan-Meier para la supervivencia global en QuANTUM-First
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con VANFLYTA en uno o más grupos de la población pediátrica en el tratamiento de la leucemia mieloide aguda (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 - Propiedades farmacocinéticas de VANFLYTA 17,7 mg Comp. recub. con película
Se evaluó la farmacocinética de quizartinib y de su metabolito activo AC886 en voluntarios adultos sanos (dosis únicas) y en pacientes con LMA de nuevo diagnóstico (en estado estacionario).
Absorción
La biodisponibilidad absoluta de quizartinib de la formulación en comprimidos fue del 71 %. Tras la administración oral en condiciones de ayuno en sujetos sanos, el tiempo hasta la concentración máxima (mediana de tmáx) de quizartinib y AC886 determinada después de administrar la dosis fue de aproximadamente 4 horas (intervalo: 2 a8 horas) y de 5 a 6 horas (intervalo: 4 a 120 horas), respectivamente.
La administración de quizartinib con alimentos, en sujetos sanos, disminuyó la Cmáx de quizartinib en
1,09 veces, aumentó el AUCinf en 1,08 veces y retrasó el tmáx dos horas. Estos cambios en la exposición no se consideran clínicamente relevantes. Se puede administrar VANFLYTA con o sin
alimentos.
Basándose en el modelo de farmacocinética poblacional en pacientes con LMA de nuevo diagnóstico a
35,4 mg/día, en estado estacionario durante la terapia de inducción, la media geométrica (% CV) de la
Cmáx de quizartinib y AC886 se estimó en 140 ng/ml (71 %) y 163 ng/ml (52 %), respectivamente, y la media geométrica (% CV) del AUC0-24h fue de 2 680 ng•h/ml (85 %) y 3 590 ng•h/ml (51 %), respectivamente.
Durante la terapia de consolidación a 35,4 mg/día, en estado estacionario, la media geométrica (% CV)
de la Cmáx de quizartinib y AC886 se estimó en 204 ng/ml (64 %) y 172 ng/ml (47 %), respectivamente, y la media geométrica (% CV) del AUC0-24h fue de 3 930 ng•h/ml (78 %) y
3 800 ng•h/ml (46 %), respectivamente.
Durante la terapia de mantenimiento a 53 mg/día, en estado estacionario, la media geométrica (% CV) de la Cmáx de quizartinib y AC886 se estimó en 529 ng/ml (60 %) y 262 ng/ml (48 %), respectivamente, y la media geométrica (% CV) del AUC0-24h fue de 10 200 ng•h/ml (75 %) y
5 790 ng•h/ml (46 %), respectivamente.
Distribución
La unión in vitro de quizartinib y AC886 a las proteínas plasmáticas humanas es superior o igual al
99 %.
El cociente sangre/plasma de quizartinib y AC886 es dependiente de la concentración, lo que indica una saturación de la distribución a los eritrocitos. A concentraciones plasmáticas clínicamente relevantes, el cociente sangre/plasma es de aproximadamente 1,3 para quizartinib y de aproximadamente 2,8 para AC886. El cociente sangre/plasma de AC886 también depende del hematocrito, con una tendencia a aumentar a niveles de hematocrito más elevados.
La media geométrica (% CV) del volumen de distribución de quizartinib en sujetos sanos se calculó en 275 l (17 %).
Biotransformación
Quizartinib se metaboliza principalmente por CYP3A4 y CYP3A5 in vitro a través de las vías oxidativas, lo que produce el metabolito activo AC886, que se metaboliza a su vez por CYP3A4 y CYP3A5. El cociente AUC0-24h entre AC886 y quizartinib en estado estacionario durante la terapia de mantenimiento fue de 0,57.
Eliminación
Las semividas efectivas (t1/2) medias (DE) de quizartinib y AC886 son de 81 horas (73) y 136 horas (113), respectivamente, en pacientes con LMA de nuevo diagnóstico. Los cocientes de acumulación (AUC0-24h) medios (DE) de quizartinib y AC886 fueron de 5,4 (4,4) y 8,7 (6,8), respectivamente.
Quizartinib y sus metabolitos se eliminan principalmente por vía hepatobiliar y la mayor parte se excreta en las heces (76,3 % de la dosis radiactiva administrada por vía oral). Quizartinib inalterado en las heces representó aproximadamente el 4 % de la dosis radiactiva administrada por vía oral. La excreción renal es una vía de eliminación menor de la dosis radiactiva administrada (<2 %).
La media geométrica (% CV) del aclaramiento corporal total (CL) de quizartinib en sujetos sanos se calculó en 2,23 l/hora (29 %).
Linealidad/No linealidad
Quizartinib y AC886 mostró una cinética lineal en el intervalo de dosis de 26,5 mg a 79,5 mg en sujetos sanos y de 17,7 mg a 53 mg en pacientes con LMA.
Relaciones farmacocinéticas/farmacodinámicas
La edad (18 a 91 años), la raza, el sexo, el peso corporal o la insuficiencia renal (CLcr de 30 a
89 ml/min, calculada mediante Cockcroft-Gault) no tuvieron ningún efecto clínicamente relevante en la exposición de quizartinib y AC886 en función del análisis de farmacocinética poblacional.
Estudios de interacciones con otros medicamentos
Transportadores
Los estudios in vitro demostraron que quizartinib es un sustrato de la P-gp, pero no de BCRP, OATP1B1, OATP1B3, OCT1, OAT2, MATE1 o MRP2. AC886 es un sustrato de la BCRP, pero no de OATP1B1, OATP1B3, MATE1 o MRP2. Sin embargo, la administración de una dosis única de quizartinib con ketoconazol, un inhibidor potente tanto de CYP3A como de P-gp, aumentó la Cmáx de quizartinib en aproximadamente 1,17 veces, lo que sugiere que el efecto de la P-gp es mínimo. Como se requiere un ajuste de dosis para el uso concomitante con inhibidores potentes de CYP3A, muchos de los cuales también inhiben la P-gp, no se requiere un ajuste de dosis específico para los inhibidores de la P-gp.
Sustratos de la proteína de resistencia al cáncer de mama (BCRP)
Quizartinib inhibe la BCRP con una CI50 in vitro estimada de 0,813 μM. Dado que no se dispone de datos clínicos, no se puede descartar que quizartinib pueda inhibir este transportador a las dosis recomendadas.
Sustratos de la uridina difosfato glucuronosiltransferasa (UGT)1A1
Quizartinib inhibe la UGT1A1 con un Ki in vitro estimado de 0,78 μM. En función de un análisis farmacocinético de base fisiológica, se predijo que quizartinib aumentaba la Cmáx y el AUCinf de raltegravir (un sustrato de UGT1A1) en 1,03 veces, lo cual no se consideró clínicamente relevante.
Poblaciones especiales
Insuficiencia hepática
En un estudio de fase 1 con dosis únicas (26,5 mg), se evaluó la farmacocinética de quizartinib y de AC886 en sujetos con insuficiencia hepática leve (clase A de Child-Pugh) o moderada (clase B de Child-Pugh) en comparación con sujetos con función hepática normal. La exposición (Cmáx y AUCinf) de quizartinib y AC886 fue similar (diferencia ≤30 %) en todos los grupos. La unión de quizartinib y AC886 a las proteínas no se ve afectada por la insuficiencia hepática. Por lo tanto, la insuficiencia hepática no tuvo ningún efecto clínicamente relevante en la exposición de quizartinib y AC886.
No se recomienda ajustar la dosis en pacientes con insuficiencia hepática leve o moderada.
Los estudios clínicos no incluyeron pacientes con insuficiencia hepática grave (clase C de Child-Pugh)
y, por lo tanto, no se recomienda el uso de VANFLYTA en estos pacientes.
Insuficiencia renal
Un análisis de farmacocinética poblacional en pacientes con LMA con insuficiencia renal leve o moderada (CLcr de 30 a 89 ml/min) mostró que la función renal no afectaba al aclaramiento de
quizartinib y AC886. Por lo tanto, la insuficiencia renal leve o moderada no tuvo ningún efecto clínicamente relevante en la exposición de quizartinib y AC886. No se recomienda ajustar la dosis en
pacientes con insuficiencia renal leve o moderada.
Los estudios clínicos no incluyeron pacientes con insuficiencia renal grave (CLcr <30 ml/min) y, por lo tanto, no se recomienda el uso de VANFLYTA en estos pacientes.
5.3 - Datos preclínicos sobre seguridad de VANFLYTA 17,7 mg Comp. recub. con película
En los estudios de genotoxicidad, quizartinib fue mutagénico en un ensayo de mutación inversa bacteriana, pero no en un ensayo de mutación celular en mamíferos (timidina quinasa de linfoma de ratón) ni en un ensayo de mutación en roedores transgénicos in vivo. Quizartinib no fue clastogénico ni indujo poliploidía en un ensayo de aberraciones cromosómicas y tampoco fue clastogénico o aneugénico en un ensayo de micronúcleos de médula ósea en ratas con dosis únicas. Un ensayo de micronúcleos de médula ósea in vivo en ratas resultó equívoco tras dosis repetidas durante 28 días. Tras una dosis única más alta, el resultado fue negativo.
No se han realizado estudios de fertilidad en animales con quizartinib. Sin embargo, se observaron hallazgos adversos en los sistemas reproductores masculinos y femeninos en los estudios de toxicidad a dosis repetidas en ratas y monos. En ratas hembra, se observaron quistes en los ovarios y modificaciones en la mucosa vaginal con dosis de aproximadamente 10 veces la dosis humana recomendada (DHR) en función del AUC. Los hallazgos en los monos hembra incluyeron atrofia uterina, ovárica y vaginal, que se observaron a dosis aproximadamente 0,3 veces la DHR en función del AUC. Los correspondientes niveles sin efecto adverso observado (NOAEL) para estos cambios fueron 1,5 veces y 0,1 veces la DHR, respectivamente, en función del AUC. En ratas macho, se observaron degeneración de los túbulos seminíferos de los testículos y ausencia de liberación de espermatozoides a dosis aproximadamente 8 veces la DHR en función del AUC. Los hallazgos en los monos macho incluyeron depleción de las células germinales en los testículos, que se observó a dosis aproximadamente 0,5 veces la DHR en función del AUC. Los correspondientes NOAEL para estos cambios fueron 1,4 veces y 0,1 veces la DHR, respectivamente, en función del AUC. Tras un periodo de recuperación de cuatro semanas, todos estos hallazgos, excepto las modificaciones de la mucosa vaginal en las ratas hembra, fueron reversibles.
En estudios de toxicidad embriofetal, se observaron mortalidad embriofetal y un aumento de las pérdidas posimplantación a dosis maternalmente tóxicas. Se observaron fetotoxicidad (menor peso fetal, efectos en la osificación esquelética) y teratogenicidad (anomalías fetales, incluido edema) a dosis aproximadamente 3 veces la DHR en función del AUC. El NOAEL fue 0,5 veces la DHR en función del AUC. Quizartinib se considera potencialmente teratogénico.
Estudios de toxicología en animales
En los estudios de toxicidad a dosis repetidas, se observó toxicidad en los órganos hematopoyéticos y linfoides, incluidas disminución de las células sanguíneas periféricas e hipocelularidad de la médula ósea; toxicidad hepática, incluidas aminotransferasas elevadas, necrosis hepatocelular y deposición de cristales birrefringentes (perros); y toxicidad renal, incluidas basofilia tubular y deposición de cristales birrefringentes (ratas macho). Estos cambios se observaron a aproximadamente 0,4 veces, 0,4 veces y
9 veces la DHR, respectivamente, en función del AUC. Los correspondientes NOAEL fueron aproximadamente 0,1 veces, 0,1 veces y 1,5 veces la DHR, respectivamente, en función del AUC.
Los estudios de evaluación del riesgo medioambiental han demostrado que quizartinib puede suponer un riesgo para el medio acuático.
Estudios de farmacología de seguridad in vitro y en animales
En los estudios de farmacología de seguridad cardiovascular realizados en monos Cynomolgus, quizartinib produjo una prolongación del QT a dosis aproximadamente 2 veces superiores a la DHR de
53 mg/día en función de la Cmáx. El NOAEL fue aproximadamente 0,4 veces la DHR en función de la
Cmáx. Quizartinib inhibió principalmente la IKs con una inhibición máxima del 67,5 % a 2,9 µM. La inhibición máxima de la IKs por AC886 fue del 26,9 % a 2,9 µM. Quizartinib y AC886, a 3 μM, inhibieron de forma estadísticamente significativa la corriente hERG en un 16,4 % y un 12,0 %, respectivamente. Ni quizartinib ni AC886 inhibieron la INa, INa-L e ICa-L a ninguna de las concentraciones probadas.
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de VANFLYTA 17,7 mg Comp. recub. con película
VANFLYTA 17,7 mg comprimidos recubiertos con película
Núcleo del comprimido
Hidroxipropilbetadex
Celulosa microcristalina (E460) Estearato de magnesio
Recubrimiento con película
Hipromelosa (E464) Talco (E553b) Triacetina (E1518)
Dióxido de titanio (E171)
VANFLYTA 26,5 mg comprimidos recubiertos con película
Núcleo del comprimido
Hidroxipropilbetadex
Celulosa microcristalina (E460) Estearato de magnesio
Recubrimiento con película
Hipromelosa (E464) Talco (E553b) Triacetina (E1518)
Dióxido de titanio (E171) Óxido de hierro amarillo E172
6.2 - Incompatibilidades de VANFLYTA 17,7 mg Comp. recub. con película
No procede.
6.3 - Período de validez de VANFLYTA 17,7 mg Comp. recub. con película
3 años.
6.4 - Precauciones especiales de conservación de VANFLYTA 17,7 mg Comp. recub. con película
No requiere condiciones especiales de conservación.
6.5 - Naturaleza y contenido del recipiente de VANFLYTA 17,7 mg Comp. recub. con película
Blísteres unidosis de aluminio/aluminio perforados. VANFLYTA 17,7 mg comprimidos recubiertos con película
Cajas que contienen 14 x 1 o 28 x 1 comprimidos recubiertos con película.
VANFLYTA 26,5 mg comprimidos recubiertos con película
Cajas que contienen 14 x 1, 28 x 1 o 56 x 1 comprimidos recubiertos con película. Puede que solamente estén comercializados algunos tamaños de envases.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de VANFLYTA 17,7 mg Comp. recub. con película
Este medicamento puede suponer un riesgo para el medio ambiente. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Daiichi Sankyo Europe GmbH Zielstattstrasse 48
81379 Munich
Alemania
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/23/1768/001-005
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 06/noviembre/2023
10. - FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia
Europea de Medicamentos https://www.ema.europa.eu.

