 OXBRYTA 500 MG COMPRIMIDOS RECUBIERTOS CON PELICULA
OXBRYTA 500 MG COMPRIMIDOS RECUBIERTOS CON PELICULA
| ATC: Voxelotor |
| PA: Voxelotor |
Envases
- Env. con 90
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica- Comercializado: No
- Situación: Alta
- Código Nacional: 764165
- EAN13: 8470007641650
- Conservar en frío: No
 Introducción
Introducción
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Oxbryta 500 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 500 mg de voxelotor. Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Comprimido recubierto con película, de color amarillo a amarillo claro, ovalado, biconvexo, de aproximadamente 18 mm × 10 mm y con la inscripción “GBT 500” en una cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Oxbryta está indicado para el tratamiento de la anemia hemolítica debida a la enfermedad de células falciformes (ECF) en adultos y pacientes pediátricos de 12 años o más, como monoterapia o en combinación con hidroxicarbamida.
4.2 Posología y forma de administración
El tratamiento lo deben iniciar médicos con experiencia en el manejo de la ECF. Posología
La dosis recomendada de Oxbryta es de 1500 mg (tres comprimidos recubiertos con película de
500 mg) por vía oral una vez al día.
Si se olvida una dosis, el tratamiento se debe continuar al día siguiente de la dosis olvidada.
Población pediátrica
La dosis recomendada de Oxbryta en pacientes de 12 a <18 años de edad es la misma que para los adultos.
No se ha establecido todavía la seguridad y eficacia de Oxbryta en pacientes pediátricos menores de
12 años. No se dispone de datos.
Poblaciones especiales
Insuficiencia renal
No se recomienda ajustar la dosis en pacientes con insuficiencia renal de leve a grave. No se ha evaluado Oxbryta en pacientes con enfermedad renal terminal (ERT) que requieren diálisis (ver
sección 4.4).
Insuficiencia hepática
No se recomienda ajustar la dosis de Oxbryta en pacientes con insuficiencia hepática leve o moderada. La dosis recomendada de voxelotor en pacientes con insuficiencia hepática grave (Child Pugh C) es de
1000 mg (dos comprimidos recubiertos con película de 500 mg) una vez al día (ver sección 4.4).
Forma de administración
Los comprimidos recubiertos con película de Oxbryta se deben tragar enteros con agua. Oxbryta se puede tomar con o sin alimentos (ver sección 5.2). Los comprimidos no se deben cortar, triturar o masticar ya que tienen un sabor desagradable.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 (ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Reacciones de hipersensibilidad
Se han observado reacciones de hipersensibilidad graves en <1 % de los pacientes tratados con voxelotor en los estudios clínicos. Las manifestaciones clínicas pueden incluir erupción cutánea generalizada, urticaria, dificultad respiratoria leve, hinchazón facial leve y eosinofilia (ver sección 4.8).
Si se producen reacciones de hipersensibilidad, se debe interrumpir la administración de voxelotor y administrar el tratamiento médico adecuado. No se debe reiniciar voxelotor en pacientes que hayan presentado estos síntomas al utilizarlo previamente.
Reacciones adversas cutáneas graves (RACG)
Se ha notificado reacción a fármacos con eosinofilia y síntomas sistémicos (DRESS, por sus siglas en inglés), también conocida como hipersensibilidad multiorgánica, relacionada con el tratamiento con Oxbryta, que puede ser potencialmente mortal o mortal (ver sección 4.8).
En el momento de hacer la prescripción, se debe advertir a los pacientes sobre los signos y síntomas, y se les debe vigilar estrechamente para detectar reacciones cutáneas. Si aparecen signos y síntomas indicativos de estas reacciones, Oxbryta se debe retirar inmediatamente y se debe considerar un tratamiento alternativo. Si el paciente presenta una reacción grave como DRESS con el uso de Oxbryta, no se debe reiniciar el tratamiento con Oxbryta en este paciente en ningún momento.
Interferencias en las pruebas de laboratorio
La administración de Oxbryta puede interferir en la medición de los subtipos de hemoglobina (Hb) (HbA, HbS y HbF) mediante cromatografía líquida de alta resolución (HPLC). Si se requiere una cuantificación precisa de las especies de Hb, la cromatografía se debe realizar cuando el paciente no haya recibido el tratamiento con Oxbryta en los 10 días inmediatamente anteriores.
Insuficiencia renal
No se observaron diferencias clínicamente significativas en la farmacocinética de voxelotor en sujetos sin ECF con insuficiencia renal de leve a grave (ver sección 5.2). No se recomienda ajustar la dosis. No se ha evaluado la seguridad de voxelotor en pacientes con ECF y ERT que requieren diálisis.
Insuficiencia hepática
Existen datos limitados sobre la seguridad de voxelotor en pacientes con ECF con diferentes grados de insuficiencia hepática. Según los datos farmacocinéticos en sujetos sin ECF, la insuficiencia hepática grave aumenta la exposición a voxelotor (ver sección 5.2). Se debe ajustar la dosis de voxelotor en pacientes con insuficiencia hepática grave (Child Pugh C) (ver sección 4.2).
Inductores potentes de CYP3A4 concomitantes
Se debe evitar el uso concomitante de inductores potentes de CYP3A4 con Oxbryta debido al riesgo de disminución de la eficacia de voxelotor (ver sección 4.5).
Genotipos de ECF
La mayoría de los pacientes (90,5 %) del estudio pivotal de fase 3 tenía ECF de genotipo HbSS (75,2 %) o HbS/β0-talasemia (15,3 %). Por tanto, los datos de seguridad y eficacia sobre otros genotipos de la ECF son limitados.
Edad avanzada
Los estudios clínicos de voxelotor no incluyeron pacientes mayores de 65 años.
Tratamiento combinado con hidroxicarbamida
Si Oxbryta se administra en combinación con hidroxicarbamida, se debe consultar la información de prescripción de hidroxicarbamida.
Efectos inmunosupresores
Voxelotor disminuyó la respuesta inmunitaria humoral a los antígenos tanto en ratas como en monos. No se puede excluir la relevancia clínica en pacientes ya inmunodeprimidos o en pacientes tratados con inmunosupresores.
Excipientes
Este medicamento contiene menos de 23 mg de sodio (1 mmol) por 1500 mg (dosis diaria); esto es, esencialmente “exento de sodio”.
4.5 Interacción con otros medicamentos y otras formas de interacción
Efecto de otros medicamentos sobre voxelotor
Inductores potentes de CYP3A4
La coadministración de inductores potentes de CYP3A4 puede disminuir la exposición de voxelotor y puede dar lugar a una reducción de la eficacia.
Se debe evitar la coadministración de voxelotor con inductores potentes de CYP3A4 (es decir, rifampicina, fenobarbital, carbamazepina, fenitoína y extracto de hierba de San Juan).
Otras interacciones estudiadas
Itraconazol (un inhibidor potente de CYP3A4), omeprazol (agente reductor de ácidos) e hidroxicarbamida no tuvieron ningún efecto sobre la farmacocinética de voxelotor.
Efecto de voxelotor sobre otros medicamentos
Sustratos de CYP3A4
Voxelotor aumentó la exposición sistémica de midazolam (un sustrato sensible de CYP3A4). El aumento observado de la exposición del sustrato de CYP3A4, midazolam, fue de 1,6 veces en sujetos sanos a una dosis subterapéutica de voxelotor (Cmáx de voxelotor observada 7,0-8,0 µg/ml y AUC
126,3-148,9 µgh/ml). Se espera que el efecto a la dosis completa de voxelotor sea mayor. Se debe evitar la coadministración de voxelotor con sustratos sensibles de CYP3A4 con un índice terapéutico
estrecho (es decir, alfentanilo, sirolimus y tacrolimus). Si el uso concomitante es inevitable, se debe considerar la reducción de la dosis del sustrato o sustratos sensibles de CYP3A4.
Sustratos de CYP2B6
Los estudios in vitro indicaron que voxelotor actúa como inhibidor e inductor de CYP2B6 (ver sección 5.2). Actualmente se desconoce la relevancia clínica, y se recomienda tener precaución al
coadministrar voxelotor con sustratos sensibles de CYP2B6 como bupropión y efavirenz.
Sustratos de CYP2C8, CYP2C9 y CYP2C19
Voxelotor es un inhibidor in vitro de CYP2C8, CYP2C9 y CYP2C19 en concentraciones sistémicas máximas. No se observó ningún cambio en las exposiciones de S-warfarina (sustrato de CYP2C9) y de
omeprazol (sustrato de CYP2C19) en voluntarios sanos a una dosis subterapéutica de voxelotor (Cmáx de voxelotor observada 7,0-8,0 µg/ml y AUC 126,3-148,9 µgh/ml). Actualmente se desconoce el efecto a la dosis completa de voxelotor. Se recomienda tener precaución al coadministrar voxelotor con sustratos sensibles de las enzimas CYP.
Interacciones farmacológicas mediadas por transportadores
Los estudios in vitro indican que voxelotor puede actuar como inhibidor de los transportadores
OATP1B1, OAT3 y MATE1 (ver sección 5.2). Por lo tanto, se recomienda tener precaución al coadministrar voxelotor con sustratos sensibles de estos transportadores, especialmente en el caso de sustratos con un índice terapéutico estrecho.
El uso concomitante de voxelotor con digoxina (un sustrato de la P-gp) no alteró la digoxina en un grado clínicamente relevante. Voxelotor no es un inhibidor de la bomba de exportación de sales biliares (BSEP). Se desconoce si voxelotor afecta a la absorción oral de los sustratos de la proteína de resistencia del cáncer de mama (BCRP).
Anticonceptivos orales y otros agentes esteroideos
No se han realizado estudios de interacción específicos con anticonceptivos orales. Sin embargo, en función de los resultados de los estudios in vitro, no se espera un impacto negativo de voxelotor sobre
la eficacia de los anticonceptivos.
Otras interacciones estudiadas
Voxelotor no alteró la exposición sistémica de la cafeína (sustrato de CYP1A2) ni de metoprolol
(sustrato de CYP2D6).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o estos son limitados relativos al uso de voxelotor en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3).
Como medida de precaución, es preferible evitar el uso de Oxbryta durante el embarazo.
Lactancia
Se desconoce si voxelotor/metabolitos se excretan en la leche materna. Los datos farmacocinéticos/toxicológicos disponibles en animales muestran que voxelotor se excreta en la leche
y posteriormente lo absorben las crías (para mayor información, ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños. Voxelotor no debe utilizarse durante la lactancia.
Fertilidad
No se dispone de datos en humanos sobre el efecto de voxelotor en la fertilidad. En ratas, se observaron efectos sobre la motilidad y la morfología del esperma. Sin embargo, estos efectos no afectaron al rendimiento reproductivo (ver sección 5.3). Se desconoce la relevancia para los humanos.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Oxbryta sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas más frecuentes incluyen cefalea (31,8 %), diarrea (22,7 %) y dolor abdominal
(22,7 %). Las reacciones adversas graves incluyen cefalea (1,1 %) e hipersensibilidad al fármaco
(1,1 %). La suspensión de forma permanente debido a una reacción adversa se produjo en el 2,3 % de los pacientes.
Se produjeron modificaciones de la dosis (reducción de la dosis o interrupción de la misma) debido a una reacción adversa en el 13,6 % de los pacientes que recibieron voxelotor en el estudio pivotal. Las reacciones adversas que requirieron una modificación de la dosis incluyeron erupción (4,5 %), diarrea (3,4 %), cefalea (2,3 %), náuseas (2,3 %), dolor abdominal (1,1 %) e hipersensibilidad al fármaco
(1,1 %).
Reacciones adversas cutáneas graves (RACG): se ha notificado reacción a fármacos con eosinofilia y síntomas sistémicos (DRESS) relacionada con el tratamiento con Oxbryta (ver sección 4.4).
Tabla de reacciones adversas
En la tabla 1 se muestran las reacciones adversas al medicamento que se produjeron en los pacientes tratados con 1500 mg de voxelotor en un estudio pivotal de fase 3, aleatorizado, doble ciego y controlado con placebo de 72 semanas (n = 88), así como las reacciones adversas durante la fase posterior a la comercialización.
Las reacciones adversas notificadas con voxelotor se enumeran de acuerdo a la clasificación por órganos y sistemas, y término preferido. Dentro de cada clasificación por órganos y sistemas, las reacciones adversas se enumeran según las categorías de frecuencia. Las frecuencias se definen como muy frecuentes (≥1/10); frecuentes (≥1/100 a <1/10); poco frecuentes (≥1/1000 a <1/100); raras
(≥1/10 000 a <1/1000); muy raras (<1/10 000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles de los estudios clínicos). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 1: Reacciones adversas
| Clasificación por órganos y sistemas | Reacciones adversasa | Categoría de frecuencia |
| Trastornos del sistema inmunológico | Hipersensibilidad al fármaco | Poco frecuentes |
| Trastornos del sistema nervioso | Cefalea | Muy frecuentes |
| Clasificación por órganos y sistemas | Reacciones adversasa | Categoría de frecuencia |
| Trastornos gastrointestinales | Diarrea Dolor abdominalb Náuseas | Muy frecuentes |
| Trastornos de la piel y del tejido subcutáneo | Erupciónc | Muy frecuentes |
| Trastornos de la piel y del tejido subcutáneo | Prurito | Frecuentes |
| Trastornos de la piel y del tejido subcutáneo | Reacción a fármacos con eosinofilia y síntomas sistémicos (DRESS) Angioedemad | Frecuencia no conocida |
a. Las reacciones adversas fueron de grado 1 o 2 del NCI, excepto la diarrea (n = 1), las náuseas (n = 1), la erupción (n = 1), la erupción generalizada (n = 3) y la hipersensibilidad (n = 1) que fueron de grado 3.
b. El dolor abdominal incluye dolor abdominal, dolor abdominal superior y dolor abdominal inferior.
c. La erupción incluye erupción, urticaria, erupción generalizada, erupción macular, erupción maculopapular, erupción prurítica y erupción papular.
d. El angioedema incluye hinchazón de los párpados, edema facial, hinchazón de los labios e hinchazón periorbitaria.
Descripción de las reacciones adversas seleccionadas
Trastornos gastrointestinales (GI)
En el estudio pivotal de fase 3, las reacciones adversas GI notificadas con mayor frecuencia fueron diarrea, dolor abdominal y náuseas, mostrando la diarrea y las náuseas un efecto dependiente de la dosis. La mayoría de las reacciones GI notificadas fueron de grado 1 o 2 y se pudieron controlar sin necesidad de interrumpir o reducir la dosis ni suspender el tratamiento, y remitieron con el uso continuado. En el 4,5 % de los pacientes se produjeron reacciones adversas gastrointestinales que conllevaron una reducción de la dosis. La diarrea fue la reacción adversa más frecuente y se notificó en el 22,7 % y el 11,0 % de los pacientes de los grupos de 1500 mg de voxelotor y placebo, respectivamente. Se notificó 1 (1,1 %) caso de diarrea de grado 3. Se produjo una reacción adversa grave de náuseas que motivó la hospitalización en 1 (1,1 %) paciente del grupo de 1500 mg de voxelotor.
Hipersensibilidad al fármaco
En el estudio pivotal de fase 3, 1 paciente (1,1 %) presentó hipersensibilidad al fármaco el día 40 del estudio. Los síntomas observados incluyeron erupción morbiliforme generalizada, urticaria, dificultad respiratoria leve, hinchazón facial leve, pirexia, cefalea y diarrea. Se observaron eosinófilos elevados. Los síntomas remitieron tras la suspensión de voxelotor, y reaparecieron tras la reintroducción de voxelotor. El episodio remitió con antihistamínicos y corticoesteroides orales.
Erupción
En el estudio pivotal de fase 3, se notificaron erupciones en el 14,8 % y el 11,0 % de los pacientes de los grupos de 1500 mg de voxelotor y placebo, respectivamente. La mayoría de las erupciones presentaban un aspecto (compatible con las típicas erupciones maculopapulares inducidas por medicamentos) y una distribución similares, no estaban asociadas a síntomas extradérmicos y se controlaron clínicamente con o sin un tratamiento que incluyera antihistamínicos orales o corticoesteroides tópicos. El análisis exposición-respuesta no reveló una relación dosis-respuesta o exposición-respuesta estadísticamente significativa.
Población pediátrica
El perfil de seguridad observado en los pacientes pediátricos de 12 a <18 años tratados con voxelotor en los estudios clínicos fue similar al observado en los pacientes adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se notificó un caso de sobredosis en el estudio pivotal de fase 3 en el que un paciente tomó un total de
3000 mg de voxelotor de una sola vez. No se observaron reacciones adversas asociadas a este episodio.
En caso de sobredosis, se debe tratar sintomáticamente al paciente y se deben tomar las medidas de soporte necesarias.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: otros agentes hematológicos, código ATC: B06AX03
Mecanismo de acción
Voxelotor es un inhibidor de la polimerización de la hemoglobina S (HbS) que se une a la HbS con una estequiometría de 1:1 y presenta una distribución preferente a los glóbulos rojos. Al aumentar la afinidad de la Hb por el oxígeno, voxelotor demuestra una inhibición de la polimerización de la HbS dependiente de la dosis. Voxelotor inhibe la formación de glóbulos rojos falciformes y mejora la deformabilidad eritrocitaria.
Efectos farmacodinámicos
El efecto farmacodinámico del tratamiento con voxelotor demostró un aumento dependiente de la dosis en la afinidad de la Hb por el oxígeno, determinada por el cambio en p20 y p50 (presión parcial de oxígeno a la que se alcanza una saturación de oxígeno de la Hb del 20 % o del 50 %) que se correlacionó linealmente con la exposición a voxelotor, lo que llevó a la inhibición de la polimerización de la HbS. El efecto de la antipolimerización es la reducción de las medidas de hemólisis (bilirrubina indirecta) con una disminución concomitante del recuento porcentual de reticulocitos y un aumento de la Hb coherente con la mejoría de la anemia hemolítica.
Electrofisiología cardiaca
En concentraciones plasmáticas aproximadamente 2 veces superiores a las concentraciones terapéuticas, voxelotor no prolonga el intervalo QT de manera clínicamente relevante.
Eficacia clínica y seguridad
Se evaluaron la eficacia y la seguridad de voxelotor en pacientes con ECF en un estudio aleatorizado, doble ciego, controlado con placebo y multicéntrico (EudraCT 2016-003370-40). En este estudio,
274 pacientes fueron aleatorizados a recibir diariamente por vía oral 1500 mg de voxelotor (N = 90),
900 mg de voxelotor (N = 92) o placebo (N = 92). Los pacientes fueron incluidos si presentaban una Hb basal de ≥5,5 g/dl (3,41 mmol/l) a ≤10,5 g/dl (6,52 mmol/l) y habían tenido de 1 a 10 episodios de crisis vasooclusivas (CVO) en los 12 meses anteriores a la inclusión. Los pacientes que, por lo demás, cumplían los requisitos y tomaban dosis estables de hidroxicarbamida durante al menos 90 días podían continuar el tratamiento con hidroxicarbamida durante todo el estudio. La aleatorización se estratificó en función de los pacientes que ya recibían hidroxicarbamida (sí, no), la región geográfica (Norteamérica, Europa, otra) y la edad (de 12 a <18 años, de 18 a 65 años). Los criterios clave de
exclusión incluyeron pacientes que (1) recibían transfusiones periódicas de glóbulos rojos, (2) habían recibido transfusiones de glóbulos rojos en los 60 días anteriores, (3) habían recibido eritropoyetina en los 28 días anteriores a la inclusión en el estudio, (4) tenían hepatitis A, B o C activa conocida o eran seropositivos al virus de la inmunodeficiencia humana (VIH), (5) tenían insuficiencia renal grave, (6) tenían una enfermedad hepática no controlada, (7) estaban embarazadas o (8) estaban en periodo de lactancia materna.
El 75 % de los pacientes tenía el genotipo HbSS, el 15 % tenía HbS/β0-talasemia, el 4 % HbS/β+- talasemia, el 3 % HbSC y el 3 % otras variantes falciformes. La mayoría recibía tratamiento con hidroxicarbamida (65 %). La mediana de edad era de 24 años (rango: 12 a 64 años); 46 (17 %) pacientes tenían entre 12 y <18 años. La mediana de Hb basal era de 8,5 g/dl (5,28 mmol/l) (5,9 a
10,8 g/dl [3,66 a 6,70 mmol/l]). Ciento quince (42 %) habían tenido 1 episodio de CVO y 159 (58 %)
habían tenido de 2 a 10 episodios en los 12 meses anteriores a la inclusión en el estudio. De los
274 pacientes, 75 (27,4 %) interrumpieron el estudio antes de tiempo. Los motivos principales de interrupción fueron la retirada del consentimiento (10,2 %) y los acontecimientos adversos (8,4 %).
La eficacia se basó en la siguiente variable primaria: tasa de respuesta de la Hb definida como un aumento de Hb de >1 g/dl (0,62 mmol/l) desde el inicio hasta la semana 24 en los pacientes tratados con 1500 mg de voxelotor frente a placebo. La tasa de respuesta de 1500 mg de voxelotor fue del
51,1 % (46/90) en comparación con el 6,5 % (6/92) en el grupo de placebo (p <0,001). No se observaron subgrupos con valores atípicos (Figura 1). Se observó un aumento de la Hb a partir de la
semana 2 y se mantuvo hasta la semana 72. La distribución del cambio en la Hb con respecto al valor
inicial para los pacientes individuales que completaron 24 semanas de tratamiento con 1500 mg de voxelotor o placebo se representa en la Figura 2.
Figura 1: Respuesta de la hemoglobina en la semana 24 por subgrupos (1500 mg de voxelotor frente a placebo) (población por intención de tratar [ITT])
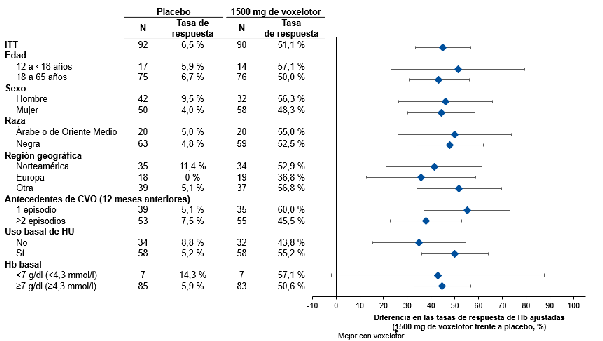
Figura 2: Cambio en la hemoglobina a nivel de sujeto desde el valor basal en la semana 24 en pacientes que completaron 24 semanas de tratamientoa,b
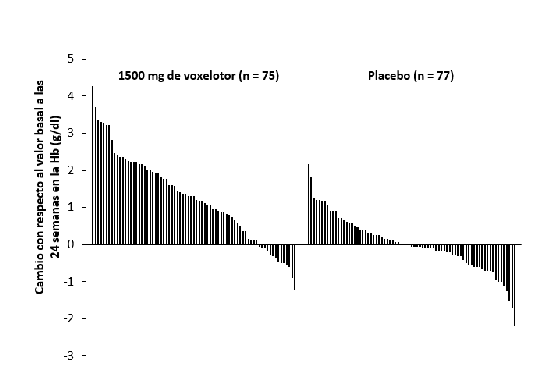
a. Aproximadamente el 83 % de todos los pacientes aleatorizados completaron 24 semanas de tratamiento.
b. En el Sistema Internacional de Unidades (SI), el rango de Hb de –3 a 5 g/dl en el eje de ordenadas (Y) equivale a –
1,86 mmol/l a 3,10 mmol/l basado en un factor de conversión de 0,6206.
La evaluación adicional de la eficacia incluyó el cambio en la Hb y el cambio porcentual en la bilirrubina indirecta y el recuento porcentual de reticulocitos desde el valor basal hasta la semana 24 y la semana 72 (Tabla 2).
Tabla 2: Cambio medio (EE) ajustado desde el valor basal hasta las semanas 24 y 72 en la hemoglobina y las medidas clínicas de hemólisis (población ITT)
| Semana 24 | Semana 72 | |||
| Oxbryta 1500 mg una vez al día (N = 90) | Placebo (N = 92) | Oxbryta 1500 mg una vez al día (N = 90) | Placebo (N = 92) | |
| Hemoglobina g/dl mmol/l | 1,13 (0,13) 0,70 (0,08) | –0,10 (0,13) –0,06 (0,08) | 1,02 (0,15) 0,63 (0,09) | 0,02 (0,15) 0,01 (0,09) |
| Valor p | <0,001 | <0,001 | ||
| Bilirrubina indirecta, % | –29,1 (3,5) | –2,8 (3,5) | –23,9 (4,9) | 2,7 (4,9) |
| Recuento porcentual de reticulocitos, % | –18,0 (4,7) | 6,8 (4,7) | –7,6 (5,5) | 11,0 (5,5) |
EE = error estándar
El número total y la tasa de incidencia (TI) anualizada de CVO durante el tratamiento fueron los siguientes: 219 episodios con una TI ajustada de 2,4 episodios/año en el grupo de 1500 mg de voxelotor y 293 episodios con una TI ajustada de 2,8 episodios/año en el grupo de placebo. No se observaron diferencias estadísticamente significativas entre los grupos de tratamiento; sin embargo, el estudio no fue diseñado para detectar una diferencia.
En el estudio pivotal se observaron úlceras en las piernas al inicio: 4 en el grupo de 1500 mg de voxelotor, 3 en el grupo de placebo. En el grupo de voxelotor, los 4 pacientes con úlceras en las piernas al inicio del estudio mejoraron después del tratamiento (en 3 pacientes remitieron en la semana 72 y en 1 paciente con una gravedad moderada al inicio del estudio pasó a ser leve). Un
paciente presentó nuevas úlceras en las piernas durante el tratamiento. Por el contrario, en el grupo de placebo, solo 1 de los 3 pacientes con úlceras en las piernas al inicio del estudio presentó una mejoría
y 5 pacientes presentaron nuevas úlceras en las piernas.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los
resultados de los ensayos realizados con voxelotor en la población pediátrica desde el nacimiento hasta
<6 meses de edad para el tratamiento de la anemia hemolítica debida a la ECF. Ver sección 4.2 para consultar la información sobre el uso en la población pediátrica.
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con voxelotor en la población pediátrica de 6 meses a <12 años de edad para el tratamiento de la anemia hemolítica debida a la ECF, así como los datos de estudios en la población pediátrica menor de 18 años. Ver sección 4.2 para consultar la información sobre el uso en
la población pediátrica.
Estudio GBT440 007
El estudio GBT440 007 es un estudio en curso de fase 2, multicéntrico, abierto y de dosis únicas y múltiples diseñado para evaluar la seguridad, la tolerabilidad, la FC y la eficacia de voxelotor en
pacientes pediátricos con ECF. Se comentan aquí los datos de eficacia y seguridad de la parte de dosis
múltiples completada en pacientes de 12 a <18 años con ECF (HbSS o HbS/β0-talasemia) que recibieron 900 mg o 1500 mg de voxelotor durante 24 semanas.
En total, 25 pacientes recibieron 900 mg de voxelotor y 15 pacientes recibieron 1500 mg de voxelotor. La mediana de edad en el grupo de 1500 mg de voxelotor era de 14 años (rango: 12-17 años), el 33 % eran chicos y el 73 % era de raza negra. La mayoría de los pacientes del grupo de 1500 mg tenía el genotipo HbSS (80 %) y todos tomaban hidroxicarbamida al inicio del estudio. El 33 % no había tenido CVO en los 12 meses anteriores a la selección y el 33 % había tenido 1 o 2 CVO en los
12 meses anteriores a la selección. La mediana del nivel basal de Hb era de 8,8 g/dl (5,46 mmol/l). El
88,0 % de los pacientes del grupo de 900 mg de voxelotor y el 80,0 % de los pacientes del grupo de
1500 mg de voxelotor completaron el estudio con 24 semanas de tratamiento. Un paciente del grupo de 1500 mg de voxelotor interrumpió el estudio debido a una reacción adversa (diarrea de grado 1).
Las evaluaciones de eficacia incluyeron medidas clínicas de anemia (Hb) y hemólisis (recuento porcentual de reticulocitos y bilirrubina indirecta). En consonancia con los resultados del estudio de fase 3 de voxelotor, se observaron mejorías en la Hb ya en la semana 2 y se mantuvieron hasta la semana 24: el cambio medio en la Hb desde el valor basal hasta la semana 20/semana 24 fue de
0,7 g/dl (0,43 mmol/l) para el grupo de 1500 mg, la disminución del recuento porcentual de reticulocitos a las 24 semanas fue de –17,4 % (–35,6, –36,5) y la disminución de la bilirrubina indirecta fue de –42,8 % (–50,5, –15,4) en el grupo de 1500 mg de voxelotor. El perfil de seguridad fue coherente con el observado en el estudio de fase 3.
5.2 Propiedades farmacocinéticas
Absorción
La mediana de Tmáx en plasma y sangre completa de voxelotor tras su administración oral es de
2 horas. Las concentraciones máximas medias en sangre completa y glóbulos rojos se observan entre 6 y 18 horas después de la administración oral. La FC es lineal en el rango de dosis de 100 mg a
2.800 mg. El estado estacionario tras la administración repetida se alcanza en 8 días y las exposiciones plasmáticas y en sangre completa de voxelotor (Tabla 3) son coherentes con la acumulación predicha en función de los datos de dosis únicas en pacientes con ECF.
Tabla 3: Parámetros farmacocinéticos de voxelotor en plasma y sangre completa (sujetos con
ECF)
| Parámetro FC | Media geométrica (%CV) de 1500 mg de voxelotor |
| FC plasmática | |
| AUC0-24h (µgh/ml) | 278 (28,4) |
| Cmáx (µg/ml) | 14 (24,5) |
| Semivida (horas) | 38,7 (30,2) |
| FC en sangre completa | |
| AUC0-24h (µgh/ml) | 3830 (33,5) |
| Cmáx (µg/ml) | 180 (31) |
Efecto de los alimentos
En sujetos sanos, la administración de una dosis única de 900 mg de Oxbryta con una comida rica en grasas dio lugar a un aumento del 45 % y del 42 % de la Cmáx y el AUC en sangre completa, respectivamente, en comparación con las condiciones de ayuno.
En los estudios clínicos, los sujetos con ECF tomaron voxelotor sin instrucciones respecto a la ingesta de alimentos y tuvieron exposiciones plasmáticas y en sangre completa de voxelotor similares a los sujetos con ECF que tomaron voxelotor después de un ayuno nocturno. La diferencia es inferior al
20 % para cualquiera de los parámetros y no se considera clínicamente significativa. Por tanto, voxelotor puede tomarse con o sin alimentos.
Distribución
Voxelotor se absorbe en el plasma y luego se distribuye predominantemente a los glóbulos rojos debido a su unión preferente a la Hb. El volumen de distribución aparente de voxelotor del compartimento central y del compartimento periférico en pacientes con ECF es de 333 l y 72,3 l en plasma, respectivamente. La unión a proteínas es del 99,8 % in vitro. La relación sangre-plasma es de aproximadamente 15:1 en pacientes con ECF.
La farmacocinética de voxelotor en sujetos sanos es diferente a la de los pacientes con ECF debido a las diferencias en la distribución sangre-plasma (relación 32:1). El volumen de distribución en sujetos sanos es de aproximadamente 754 l.
Biotransformación
Los estudios in vitro e in vivo indican que voxelotor se metaboliza en gran medida a través del metabolismo de fase I (oxidación y reducción), de fase II (glucuronidación) y combinaciones de fase I y II. La oxidación de voxelotor está mediada principalmente por CYP3A4, con una contribución menor de CYP2C19, CYP2B6 y CYP2C9. La sulfatación de voxelotor está mediada principalmente por SULT1B1 y SULT1C4 y la glucuronidación directa de voxelotor está mediada por UGT1A1 y UGT1A9. El metabolito plasmático principal es consecuencia de la O-desalquilación-sulfatación y representa el 16,8 % del material relacionado con voxelotor en el plasma. Otros cinco metabolitos representaron un total del 23 % del material relacionado con voxelotor en el plasma, con contribuciones individuales de hasta el 9 %. Todos los demás metabolitos representaron menos del
5 %.
Eliminación
La vía principal de eliminación de voxelotor es por metabolismo con la posterior excreción de los metabolitos en la orina y las heces. La excreción de voxelotor inalterado es mínima (<1 % de la dosis en orina). La media geométrica (%CV) de la semivida de eliminación terminal de voxelotor en pacientes con ECF es de 38,7 horas (30,2 %), con concentraciones plasmáticas y en sangre completa
que disminuyen paralelamente. El aclaramiento oral aparente de voxelotor se estimó en 6,1 l/h en plasma en pacientes con ECF.
Poblaciones especiales
Pacientes con insuficiencia renal
No hubo ningún efecto clínicamente significativo de la función renal sobre la excreción de voxelotor en sujetos sin ECF y en pacientes con ECF. Tras una dosis única de 900 mg de voxelotor, la exposición en sangre completa en los sujetos con insuficiencia renal grave (FGe <30 ml/min/1,73 m2) fue un 25 % menor en comparación con los controles sanos. Las concentraciones no unidas al plasma fueron comparables. En los pacientes con ECF, se observó una tendencia a una mayor exposición a voxelotor con niveles más bajos de cistatina C. Los niveles más elevados de cistatina C observados normalmente en la insuficiencia renal no se asociaron a una mayor exposición a voxelotor.
Voxelotor no se ha evaluado en pacientes con enfermedad renal terminal que requieren diálisis.
Pacientes con insuficiencia hepática
En plasma, la Cmáx fue 1,2 veces mayor en sujetos con insuficiencia hepática leve (Child Pugh A),
1,5 veces mayor en sujetos con insuficiencia hepática moderada (Child Pugh B) y 1,4 veces mayor en sujetos con insuficiencia hepática grave (Child Pugh C), y el AUCinf fue 1,1 veces mayor en sujetos con insuficiencia hepática leve, 1,2 veces mayor en sujetos con insuficiencia hepática moderada y
1,9 veces mayor en sujetos con insuficiencia hepática grave. En sangre completa, el aumento de la exposición fue similar al del plasma. No se justifica un ajuste de la dosis en sujetos con insuficiencia
hepática de leve a moderada, pero se recomienda reducir la dosis diaria de voxelotor a 1000 mg en sujetos con insuficiencia hepática grave (ver sección 4.2). Se espera que los valores de Cmáx en plasma
y sangre completa en pacientes con insuficiencia hepática grave tras el ajuste de la dosis sean similares a los de los pacientes con función hepática normal tratados con la dosis recomendada de 1500 mg
diarios. Se espera que el AUC en plasma y sangre completa sea un ~25 % mayor en sujetos con insuficiencia hepática grave tras el ajuste de la dosis en comparación con los pacientes con función hepática normal tratados a la dosis recomendada de 1500 mg diarios.
Efecto del sexo, la raza y el peso corporal
No se observaron diferencias clínicamente significativas en la farmacocinética de voxelotor en función del sexo, la raza y el peso corporal (28 a 135 kg)
Efecto de la edad
No se observaron diferencias clínicamente significativas en la farmacocinética de voxelotor en función de la edad (12 a 59 años).
Efecto del hematocrito
La distribución de voxelotor en sangre-plasma aumenta con el incremento del hematocrito. A medida que el hematocrito aumentó del 30,5 % en los pacientes con ECF (mediana a 1500 mg diarios) al hematocrito máximo medido a 1500 mg diarios (35,1 %), la distribución sangre-plasma aumentó del
14,8 al 16,4 (aumento del 11 %).
Pacientes con genotipo HbSC
El AUC y la Cmáx de voxelotor en sangre completa en estado estacionario fueron un 50 % y un 45 %
mayores en los pacientes con genotipo HbSC (n = 11) en comparación con los pacientes con genotipo HbSS (n = 220), y el AUC y la Cmáx de voxelotor en plasma en estado estacionario fueron un 23 % y un 15 % mayores en los pacientes con genotipo HbSC en comparación con los pacientes con genotipo HbSS.
Interacciones farmacológicas in vitro
Enzimas CYP: in vitro, voxelotor es un inhibidor e inductor de CYP2B6, y un inhibidor de CYP2C8, CYP2C9, CYP2C19 y CYP3A4. Se desconoce actualmente su relevancia clínica (ver sección 4.5).
Enzimas UGT: los datos in vitro indican que voxelotor no es un inhibidor de UGT1A1, UGT1A9 y UGT2B7 a la concentración sistémica máxima. Debido a problemas de solubilidad, no se pudieron investigar concentraciones hasta la máxima concentración intestinal para UGT1A1. No se observó ninguna inhibición de UGT1A1 hasta 100 micromoles (la concentración más alta investigada).
Interacciones mediadas por transportadores: voxelotor no es un inhibidor de la P-gp, la BCRP, la OATP1B3, la OCT2, la OAT1, la MATE2-K o la BSEP. Voxelotor actúa como un inhibidor de los transportadores OATP1B1, OAT3 y MATE1 (ver sección 4.5). Voxelotor no es un sustrato de P-gp, BCRP, OATP1A2, OATP1B1, OATP1B3 o BSEP.
5.3 Datos preclínicos sobre seguridad
Las reacciones adversas no observadas en ensayos clínicos, pero detectadas en animales con niveles de exposición similares a los clínicos y con posible repercusión en el uso clínico fueron las siguientes:
Toxicidad a dosis repetidas
El hallazgo principal asociado con la administración a dosis repetidas de voxelotor fue la eritropoyesis compensatoria, manifestada como un aumento de la masa de glóbulos rojos (↑ GR, HCT, Hb, RET) correlacionado microscópicamente con una médula ósea y una pulpa roja esplénica hipercelulares y un aumento del peso esplénico en ratas, ratones y monos cynomolgus. En los monos, las fases tempranas de este efecto se observaron a niveles de dosis comparables con la exposición clínica (múltiplo de exposición de ~0,6 basado en los valores de Cmáx en plasma). Voxelotor también causó intolerancia GI atribuida a la irritación local. Otros hallazgos atribuidos a voxelotor incluyen la inducción de enzimas CYP en el hígado de ratones y ratas, la alteración de la respuesta antigénica dependiente de células T en roedores y monos y la prolongación de los intervalos QT corregidos (QTc) en monos. Tras la inmunización con hemocianina de lapa de ojo de cerradura (KLH), voxelotor produjo una reducción significativa de los títulos de IgG (ratas, monos) e IgM (monos), un retraso en el nivel máximo de la respuesta de anticuerpos (monos) y cambios en la distribución relativa de los linfocitos (ratas). Estos efectos se observaron en el múltiplo de exposición de la exposición clínica prevista de ~0,6 en los monos y ~4,0 en las ratas según el valor de la Cmáx del plasma. El tratamiento con voxelotor en el múltiplo de exposición de ~2,5 de la exposición clínica prevista produjo una prolongación de los intervalos QT y QTc en los monos.
Reproducción y desarrollo
El tratamiento de ratas con voxelotor a un múltiplo de exposición de ~4 de la exposición clínica prevista produjo una reducción de la motilidad de los espermatozoides y un aumento del porcentaje de espermatozoides anormales, así como un aumento del peso testicular y de la próstata y una reducción del peso de las vesículas seminales. Sin embargo, estos efectos no afectaron al rendimiento reproductivo. Voxelotor no fue teratogénico en ratas y conejos a los niveles de exposición que producen toxicidad materna (múltiplo de exposición basado en el AUC en sangre de 2,8 en ratas y 0,3 en conejos). Voxelotor se excreta en la leche de ratas lactantes. La exposición a través de la leche fue hasta 0,4 veces superior a la exposición plasmática de las madres, lo que llevó a la posterior
exposición plasmática de las crías. En el estudio de toxicidad en el desarrollo prenatal y posnatal, se observaron efectos adversos en la descendencia, manifestados como un índice de viabilidad de las
crías reducido y un peso persistentemente inferior de las mismas, en el múltiplo de exposición previsto
de ~2,6 de la exposición humana prevista.
Evaluación del riesgo medioambiental (ERA, por sus siglas en inglés)
Los estudios de evaluación del riesgo medioambiental han demostrado que voxelotor no es bioacumulable ni tóxico para el medio ambiente; sin embargo, tiene el potencial de persistir en los sedimentos (ver sección 6.6).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido
Celulosa microcristalina (E 460) Croscarmelosa sódica (E 468) Laurilsulfato de sodio (E 487) Sílice coloidal anhidra (E 551) Estearato de magnesio (E 470b)
Recubrimiento con película del comprimido
Alcohol polivinílico (E 1203) Dióxido de titanio (E 171) Polietilenglicol (E 1521) Talco (E 553b)
Óxido de hierro amarillo (E 172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Frasco de polietileno de alta densidad (HDPE) con una cápsula de cierre de polipropileno a prueba de niños y un precinto de inducción de aluminio. El frasco también contiene un bote de desecante de gel de sílice y una espiral de poliéster.
Envase de 90 comprimidos recubiertos con película.
6.6 Precauciones especiales de eliminación
Este medicamento puede persistir en el medio ambiente (ver sección 5.3).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Bélgica
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/21/1622/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 14/febrero/2022
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ANEXO II
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Global Blood Therapeutics Netherlands B.V. Strawinskylaan 3051
1077ZX Amsterdam
Países Bajos
O
Pfizer Service Company BV Hoge Wei 10
1930 Zaventem
Bélgica
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las
Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Informes periódicos de seguridad (IPSs)
Los requerimientos para la presentación de los IPSs para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater,
apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
El titular de la autorización de comercialización (TAC) presentará el primer IPS para este medicamento en un plazo de 6 meses después de la autorización.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Plan de gestión de riesgos (PGR)
El titular de la autorización de comercialización (TAC) realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el
Módulo 1.8.2 de la autorización de comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
A petición de la Agencia Europea de Medicamentos.
Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ANEXO III ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
CAJA EXTERIOR

1. NOMBRE DEL MEDICAMENTO
![]()
![]()
Oxbryta 500 mg comprimidos recubiertos con película voxelotor
2. PRINCIPIO(S) ACTIVO(S)
![]()
Cada comprimido recubierto con película contiene 500 mg de voxelotor.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
![]()
![]()
90 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
![]()
Leer el prospecto antes de utilizar este medicamento. Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
![]()
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
![]()
![]()
CAD

9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO, CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
Pfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
![]()
Bélgica
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
EU/1/21/1622/001
13. NÚMERO DE LOTE
![]()
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
![]()
16. INFORMACIÓN EN BRAILLE
![]()
![]()
Oxbryta 500 mg
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
![]()
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC SN NN
INFORMACIÓN QUE DEBE FIGURAR EN EL ACONDICIONAMIENTO PRIMARIO
ETIQUETA DEL FRASCO

1. NOMBRE DEL MEDICAMENTO
![]()
![]()
Oxbryta 500 mg comprimidos recubiertos con película voxelotor
2. PRINCIPIO(S) ACTIVO(S)
![]()
Cada comprimido recubierto con película contiene 500 mg de voxelotor.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
![]()
![]()
90 comprimidos recubiertos con película.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
![]()
Leer el prospecto antes de utilizar este medicamento. Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
![]()
Mantener fuera de la vista y del alcance de los niños. No tragar el desecante.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
![]()
![]()
CAD

9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO, CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
Pfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
![]()
Bélgica
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
EU/1/21/1622/001
13. NÚMERO DE LOTE
![]()
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
![]()
16. INFORMACIÓN EN BRAILLE
![]()
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
![]()
![]()
Incluido el código de barras 2D que lleva el identificador único en la caja.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
B. PROSPECTO
Prospecto: información para el paciente
Oxbryta 500 mg comprimidos recubiertos con película
voxelotor
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Oxbryta y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Oxbryta
3. Cómo tomar Oxbryta
4. Posibles efectos adversos
5. Conservación de Oxbryta
6. Contenido del envase e información adicional
1. Qué es Oxbryta y para qué se utiliza
Qué es Oxbryta y cómo funciona
Oxbryta contiene el principio activo voxelotor. Voxelotor actúa sobre una proteína de los glóbulos rojos llamada hemoglobina para ayudarla a captar el oxígeno que los glóbulos rojos pueden suministrar por todo el cuerpo.
Los pacientes que padecen la enfermedad llamada enfermedad de las células falciformes presentan una forma alterada de hemoglobina, llamada hemoglobina falciforme, que es diferente de la hemoglobina normal. Cuando la hemoglobina falciforme suministra el oxígeno a los tejidos, se adhiere formando largos filamentos y hace que los glóbulos rojos alteren su forma a la de una luna creciente, haciendo que estas células sean rígidas y tengan forma de hoz. Los glóbulos rojos falciformes no pueden suministrar el oxígeno como lo hacen los glóbulos rojos sanos y, además, se descomponen más rápidamente, lo que provoca una disminución de los niveles de glóbulos rojos (anemia hemolítica). Al mejorar la forma en que la hemoglobina alterada retiene el oxígeno, Oxbryta mejora la función de los glóbulos rojos y prolonga su vida.
Para qué se utiliza Oxbryta
Oxbryta, solo o junto con hidroxicarbamida (también conocida como hidroxiurea), se utiliza para tratar la anemia hemolítica en adultos y niños a partir de 12 años con enfermedad de las células falciformes.
2. Qué necesita saber antes de empezar a tomar Oxbryta
No tome Oxbryta
Si es alérgico a voxelotor o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar Oxbryta si tiene:
problemas renales graves;
problemas hepáticos graves. Es posible que su médico necesite ajustarle la dosis de Oxbryta.
Si presenta algún síntoma de una reacción alérgica, deje de tomar Oxbryta y consulte a su médico o acuda a urgencias inmediatamente. Los síntomas son, por ejemplo, erupción, incluidas ronchas (urticaria), dificultad para respirar e hinchazón de la cara.
Se ha notificado una reacción cutánea grave, como la reacción a fármacos con eosinofilia y síntomas sistémicos (DRESS, por sus siglas en inglés), relacionada con el tratamiento con Oxbryta. Deje de usar Oxbryta y solicite asistencia médica inmediatamente si nota alguno de los síntomas relacionados con esta reacción cutánea grave que se describen en la sección 4.
Si está recibiendo transfusiones de sangre, consulte a su médico sobre las posibles dificultades en la interpretación de ciertos análisis de sangre cuando tome este medicamento.
Niños menores de 12 años
Este medicamento no está recomendado para niños menores de 12 años debido a la falta de datos en este grupo de edad.
Otros medicamentos y Oxbryta
Informe a su médico si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento.
Algunos medicamentos pueden afectar al modo de actuar de Oxbryta o hacer que los efectos adversos sean más probables. En concreto, informe a su médico si toma alguno de los siguientes medicamentos:
rifampicina (utilizada para tratar las infecciones bacterianas);
fenobarbital, carbamazepina, fenitoína (utilizados para tratar la epilepsia y otras enfermedades);
sirolimus, tacrolimus (utilizados para prevenir el rechazo de órganos después de un trasplante);
hierba de san Juan (un medicamento a base de plantas para tratar la depresión);
alfentanilo (un analgésico utilizado durante una operación con anestesia).
Informe a su médico de que está tomando Oxbryta si va a someterse a una intervención médica o quirúrgica.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Embarazo
Su médico le ayudará a decidir si debe dejar de tomar Oxbryta durante el embarazo.
Lactancia
No dé el pecho mientras tome Oxbryta porque se desconoce si voxelotor pasa a la leche materna y puede afectar al bebé.
Conducción y uso de máquinas
La influencia de Oxbryta sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
Oxbryta contiene sodio
Este medicamento contiene menos de 23 mg de sodio (1 mmol) por dosis de tres comprimidos; esto es, esencialmente “exento de sodio”.
3. Cómo tomar Oxbryta
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
La dosis recomendada para adultos y niños a partir de 12 años es:
Tres comprimidos de 500 mg tomados una vez al día por vía oral.
Trague los comprimidos enteros con un vaso de agua, con o sin alimentos. No corte, triture o mastique los comprimidos ya que saben mal.
Si toma más Oxbryta del que debe
Póngase en contacto con su médico inmediatamente.
Si olvidó tomar Oxbryta
Continúe con su horario de administración normal al día siguiente. No tome una dosis doble para compensar las dosis olvidadas.
Si interrumpe el tratamiento con Oxbryta
No deje de tomar este medicamento sin consultar a su médico. Es importante que tome Oxbryta a diario.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Deje de tomar Oxbryta e informe a su médico o acuda a urgencias inmediatamente si presenta alguno de los siguientes efectos adversos graves:
Poco frecuentes (pueden afectar hasta a 1 de cada 100 personas)
reacciones alérgicas
Los síntomas son, por ejemplo, erupción, incluidas ronchas (urticaria), dificultad para respirar e hinchazón de la cara.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
erupción generalizada, temperatura corporal alta y ganglios linfáticos agrandados (síndrome
DRESS o síndrome de hipersensibilidad a fármacos)
hinchazón de los párpados, cara y labios (angioedema)
Pueden producirse otros efectos adversos con la siguiente frecuencia:
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
dolor de cabeza
diarrea
dolor abdominal (de estómago)
náuseas
erupción
Frecuentes (pueden afectar hasta a 1 de cada 10 personas)
picor (prurito)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Oxbryta
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el frasco y la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
Este medicamento no requiere condiciones especiales de conservación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Oxbryta
El principio activo es voxelotor. Un comprimido contiene 500 mg de voxelotor.
Los demás componentes son:
- celulosa microcristalina (E 460)
- croscarmelosa sódica (E 468)
- laurilsulfato de sodio (E 487)
- sílice coloidal anhidra (E 551)
- estearato de magnesio (E 470b)
- alcohol polivinílico (E 1203)
- dióxido de titanio (E 171)
- polietilenglicol (E 1521)
- talco (E 553b)
- óxido de hierro amarillo (E 172)
Aspecto del producto y contenido del envase
Comprimidos recubiertos con película, de color amarillo a amarillo claro, ovalados, biconvexos y con la inscripción “GBT 500” en una cara. Tamaño de los comprimidos: aproximadamente
18 mm × 10 mm.
Oxbryta se presenta en un frasco de plástico con una cápsula de cierre a prueba de niños. Cada frasco contiene 90 comprimidos recubiertos con película. El frasco también contiene una espiral y un bote de desecante de gel de sílice para mantener el medicamento seco. El frasco se suministra dentro de una caja.
Titular de la autorización de comercialización
Pfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Bélgica
Responsable de la fabricación
Global Blood Therapeutics Netherlands B.V. Strawinskylaan 3051
1077ZX Amsterdam
Países Bajos
O
Pfizer Service Company BV Hoge Wei 10
1930 Zaventem
Bélgica
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización
België/Belgique/Belgien Luxembourg/Luxemburg Pfizer NV/SA
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel: +370 5 251 4000
България
Пфайзер Люксембург САРЛ, Клон България
Тел.: +359 2 970 4333
Magyarország
Pfizer Kft.
Tel.: +36 1 488 37 00
Česká republika
Pfizer, spol. s r.o.
Tel: +420 283 004 111
Malta
Vivian Corporation Ltd. Tel: +356 21344610
Danmark
Pfizer ApS
Tlf: +45 44 20 11 00
Nederland
Pfizer bv
Tel: +31 (0)800 63 34 636
Deutschland
PFIZER PHARMA GmbH Tel: +49 (0)30 550055-51000
Norge
Pfizer AS
Tlf: +47 67 52 61 00
Eesti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Österreich
Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
Ελλάδα
Pfizer Ελλάς A.E.
Τηλ: +30 210 6785800
Polska
Pfizer Polska Sp. z o.o., Tel.: +48 22 335 61 00
España
Pfizer, S.L.
Tel: +34 91 490 99 00
Portugal
Laboratórios Pfizer, Lda. Tel: +351 21 423 5500
France
Pfizer
Tél: +33 (0)1 58 07 34 40
România
Pfizer Romania S.R.L. Tel: +40 (0) 21 207 28 00
Hrvatska
Pfizer Croatia d.o.o. Tel: + 385 1 3908 777
Slovenija
Pfizer Luxembourg SARL
Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana
Tel: + 386 (0)1 52 11 400
Ireland
Pfizer Healthcare Ireland
Tel: +1800 633 363 (toll free) Tel: +44 (0)1304 616161
Slovenská republika
Pfizer Luxembourg SARL, organizačná zložka
Tel: + 421 2 3355 5500
Ísland
Icepharma hf.
Sími: +354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
Italia
Pfizer S.r.l.
Tel: +39 06 33 18 21
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
Κύπρος
Pfizer Ελλάς Α.Ε. (Cyprus Branch) Τηλ: +357 22817690
United Kingdom (Northern Ireland)
Pfizer Limited
Tel: +44 (0)1304 616161
Latvija
Pfizer Luxembourg SARL filiāle Latvijā
Tel: +371 670 35 775
Fecha de la última revisión de este prospecto: MM/AAAA. Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
ANEXO IV
CONCLUSIONES CIENTÍFICAS Y MOTIVOS PARA LA MODIFICACIÓN DE LAS CONDICIONES
DE LAS AUTORIZACIONES DE COMERCIALIZACIÓN
Conclusiones científicas
Teniendo en cuenta lo dispuesto en el Informe de Evaluación del Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC) para voxelotor, las conclusiones científicas del PRAC son las siguientes:
En vista de los datos disponibles sobre los riesgos procedentes de los ensayos clínicos y las notificaciones espontáneas que incluyen en algunos casos una relación temporal estrecha, una retirada positiva o una nueva exposición, el PRAC considera que una relación causal entre voxelotor y prurito, así como entre voxelotor y angioedema (que incluye hinchazón de los labios, hinchazón de los párpados, edema facial e hinchazón periorbitaria) es, como mínimo, una posibilidad razonable. El PRAC concluyó que la información de los medicamentos que contienen voxelotor se debe modificar en consecuencia.
Tras estudiar la recomendación del PRAC, el CHMP está de acuerdo con las conclusiones generales del PRAC y con los motivos para la recomendación.
Motivos para la modificación de las condiciones de la(s) autorización(es) de comercialización
De acuerdo con las conclusiones científicas para voxelotor, el CHMP considera que el balance beneficio-riesgo del medicamento o medicamentos que contiene(n) voxelotor no se modifica sujeto a los cambios propuestos en la información del producto.
El CHMP recomienda que se modifiquen las condiciones de la(s) autorización(es) de comercialización.

