LYFNUA Comprimido
| ATC: Gefapixant |
| PA: Gefapixant Citrato |
Envases
 Introducción
Introducción
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Lyfnua 45 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene citrato de gefapixant equivalente a 45 mg de gefapixant.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido)
Comprimido de color rosa, de 10 mm, redondo y convexo, grabado con “777” en una cara y liso en la otra.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lyfnua está indicado en adultos para el tratamiento de la tos crónica refractaria o idiopática.
4.2 Posología y forma de administración
Posología
La dosis recomendada de gefapixant es de un comprimido de 45 mg por vía oral dos veces al día con o sin alimentos.
Dosis olvidadas
Se debe indicar a los pacientes que, si olvidan una dosis, deben saltársela y continuar con la pauta habitual. Los pacientes no deben tomar una dosis doble en su siguiente toma ni tomar una dosis mayor a la prescrita.
Poblaciones especiales
Pacientes de edad avanzada (≥ 65 años)
No es necesario ajustar la dosis en pacientes de edad avanzada (ver las secciones 5.1 y 5.2).
Se sabe que gefapixant se excreta de manera principal por el riñón. Debido a que es más probable que los pacientes de edad avanzada tengan una función renal disminuida, el riesgo de reacciones adversas a gefapixant puede ser mayor en estos pacientes. Se debe tener cuidado con la frecuencia de administración al inicio.
Insuficiencia renal
Es necesario ajustar la dosis en pacientes con insuficiencia renal grave (TFGe <30 ml/minuto/1,73 m2)
que no estén en diálisis. Se debe reducir la dosis a un comprimido de 45 mg una vez al día. No es necesario ajustar la dosis en pacientes con insuficiencia renal leve o moderada (TFGe
≥ 30 ml/minuto/1,73 m2). No se dispone de datos suficientes para hacer recomendaciones posológicas
en pacientes con enfermedad renal terminal que estén en diálisis (ver sección 5.2).
Insuficiencia hepática
No se han realizado estudios en pacientes con insuficiencia hepática. Sin embargo, dado que el metabolismo hepático es una vía secundaria de eliminación de gefapixant, no se recomienda ajustar la dosis (ver sección 5.2).
Población pediátrica
No es apropiado el uso de Lyfnua en la población pediátrica (menores de 18 años de edad) para la indicación de tos crónica refractaria o idiopática.
Forma de administración
Vía oral.
Los comprimidos se deben tragar enteros y se pueden tomar con o sin alimentos. Se debe indicar a los pacientes que no partan, machaquen ni mastiquen los comprimidos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Apnea obstructiva del sueño
En pacientes con apnea obstructiva del sueño (AOS, n=19) de moderada a grave que no estaban utilizando presión positiva sobre las vías respiratorias, la administración diaria de una dosis de 180 mg de gefapixant a la hora de dormir se asoció con una media de la saturación de oxígeno (SaO2) más baja y una media de la proporción de tiempo con SaO2 < 90 % más alta en todas las etapas del sueño en comparación con placebo. Se desconoce la importancia clínica de estos hallazgos en el uso de 45 mg
de gefapixant dos veces al día en pacientes con tos crónica refractaria (TCR) o tos crónica idiopática (TCI) con AOS comórbida. En pacientes con AOS, se debe valorar el tratamiento adecuado de la AOS antes de iniciar el tratamiento con gefapixant.
Hipersensibilidad
Gefapixant contiene una fracción sulfonamida pero se considera que no es una sulfonilarilamina. No se ha estudiado gefapixant en pacientes con antecedentes de hipersensibilidad a las sulfonamidas, por lo tanto, no se puede excluir una hipersensibilidad cruzada con hipersensibilidad a las sulfonamidas. Gefapixant se debe utilizar con precaución en pacientes con hipersensibilidad conocida a las sulfonamidas.
Infección aguda de la vía respiratoria inferior
El tratamiento con gefapixant se debe evaluar e individualizar en pacientes que desarrollen una infección aguda de la vía respiratoria inferior (ver sección 5.1).
Reacciones adversas relacionadas con el gusto
En los estudios clínicos se notificaron de manera muy frecuente reacciones adversas relacionadas con el gusto. En la mayoría de los pacientes, estas reacciones adversas se resolvieron poco después de suspender el tratamiento con gefapixant (mediana de tiempo de 5 días). En pocos pacientes, estas reacciones persistieron durante más de un año después de la suspensión del tratamiento (ver
sección 4.8).
Excipientes
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por comprimido; esto es, esencialmente “exento de sodio”.
4.5 Interacción con otros medicamentos y otras formas de interacción
Basado en los estudios in vitro (ver sección 5.2), se realizaron estudios apropiados de interacción clínica y no se identificaron interacciones clínicamente importantes.
Población pediátrica
Los estudios de interacción se han realizado sólo en adultos.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos relativos al uso de gefapixant en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3). Como medida de precaución, es preferible evitar el uso de Lyfnua durante el embarazo y en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
Los datos farmacodinámicos/toxicológicos disponibles en animales muestran que gefapixant se excreta en la leche (ver sección 5.3).
No se puede excluir el riesgo en recién nacidos/niños.
Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con Lyfnua tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No se dispone de datos en humanos relativos al efecto de gefapixant sobre la fertilidad. En ratas, no hubo ningún efecto sobre el apareamiento o la fertilidad con el tratamiento con gefapixant (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de gefapixant sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. En casos individuales, se puede producir mareo después de la administración de gefapixant que puede influir en la capacidad de conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas notificadas con más frecuencia fueron disgeusia (41 %), ageusia (15 %) e hipogeusia (11 %).
Tabla de reacciones adversas
La seguridad de gefapixant se evaluó en dos estudios clínicos de fase III (COUGH-1 y COUGH-2) que incluyeron un total de 1.369 pacientes tratados con gefapixant (15 mg o 45 mg dos veces al día) (ver sección 5.1). La duración de la exposición con gefapixant fue de 52 semanas.
Las reacciones adversas notificadas con gefapixant obtenidas de los estudios clínicos se enumeran en la tabla siguiente mediante la clasificación por órganos y sistemas de MedDRA y por frecuencia. Las
frecuencias se definen como: muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes
(≥1/1.000 a <1/100), raras (≥1/10.000 a <1/1.000) y muy raras (<1/10.000).
Tabla 1: reacciones adversas
| Clasificación por órganos y sistemas | Reacciones adversas |
| Infecciones e infestaciones | |
| Frecuentes | Infección del tracto respiratorio superior |
| Trastornos del metabolismo y de la nutrición | |
| Frecuentes | Apetito disminuido |
| Trastornos del sistema nervioso | |
| Muy frecuentes | Disgeusia*, Ageusia, Hipogeusia |
| Frecuentes | Trastorno del gusto, Mareo |
| Trastornos respiratorios, torácicos y mediastínicos | |
| Frecuentes | Tos**, Dolor orofaríngeo |
| Trastornos gastrointestinales | |
| Frecuentes | Náuseas, Diarrea, Boca seca, Hipersecreción salival, Dolor en la zona superior del abdomen, Dispepsia, Hipoestesia oral, Parestesia oral |
| Trastornos psiquiátricos | |
| Frecuentes | Insomnio |
| Trastornos renales y urinarios | |
| Poco frecuentes | Cálculo urinario, Nefrolitiasis, Cálculo en la vejiga |
*La disgeusia se notificó de manera frecuente como sabor amargo, sabor metálico o sabor salado.
**La tos incluye informes de "empeoramiento", "exacerbación", “intensificación” o "aumento" de la tos.
Descripción de reacciones adversas seleccionadas
Reacciones adversas relacionadas con el gusto
La mayoría de los pacientes con reacciones adversas relacionadas con el gusto (disgeusia, ageusia, hipogeusia y trastorno del gusto) experimentaron la aparición de las reacciones adversas dentro de los
9 días posteriores al comienzo del tratamiento con gefapixant; la mayoría fueron de intensidad leve (65 %) a moderada (32 %). Se produjo la resolución de las reacciones adversas relacionadas con el gusto en el 96 % de los pacientes con un 25 % que notificaron la resolución durante o antes de la última dosis de gefapixant. Las reacciones adversas relacionadas con el gusto persistieron durante más de un año después de la interrupción del tratamiento en el 1,6 % (7/447) de los pacientes del grupo de gefapixant y en el 12,8 % (6/47) de los pacientes del grupo placebo. Las reacciones adversas que dieron lugar a la suspensión del tratamiento se produjeron en el 22 % de los pacientes que recibieron gefapixant. Las reacciones adversas notificadas con más frecuencia que dieron lugar a la suspensión del tratamiento fueron disgeusia (9 %) y ageusia (4 %).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los
profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
En un estudio clínico con 8 voluntarios sanos que recibieron gefapixant 1.800 mg dos veces al día
(40 veces la dosis recomendada en humanos) durante un máximo de 14 días, se detectaron cristales en la orina de los participantes que contenían gefapixant. No se observaron pruebas de lesiones en el sistema renal o urinario.
En los casos de sobredosis notificados durante los estudios de fase III, no se notificaron acontecimientos adversos.
En caso de sobredosis se debe vigilar al paciente por si presenta reacciones adversas e instaurar medidas de apoyo adecuadas. Gefapixant se elimina parcialmente mediante hemodiálisis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros supresores de la tos, código ATC: R05DB29
Mecanismo de acción
Gefapixant es un antagonista selectivo del receptor P2X3. Gefapixant también tiene actividad frente al subtipo de receptor P2X2/3. Los receptores P2X3 son canales de iones regulados por el ATP que se encuentran en las fibras C sensitivas del nervio vago en las vías respiratorias. Las fibras C se activan en respuesta a la inflamación o a irritantes químicos. El ATP se libera de las células de la mucosa de las vías respiratorias en condiciones de inflamación. La unión del ATP extracelular a los receptores P2X3 se detecta como una señal de daño por parte de las fibras C. La activación de las fibras C, que el paciente siente como una necesidad de toser, inicia un reflejo de tos. El bloqueo de la señal que el
ATP produce a través de los receptores P2X3 reduce la activación excesiva de los nervios sensitivos y la tos excesiva inducida por el ATP extracelular.
Eficacia clínica y seguridad
Se estudió la eficacia de Lyfnua para el tratamiento de la tos crónica refractaria o idiopática en dos estudios de 52 semanas, multicéntricos, aleatorizados, doble ciego, controlados con placebo en adultos con tos crónica refractaria o idiopática. Se definió la tos crónica refractaria (TCR) como la tos
asociada a una enfermedad comórbida (p. ej., asma, enfermedad por reflujo gastroesofágico o síndrome de tos de las vías respiratorias superiores) que persistía a pesar del tratamiento adecuado de la enfermedad comórbida. Se definió la tos crónica idiopática (TCI) como la tos que no se asociaba a ninguna enfermedad comórbida a pesar de una evaluación clínica exhaustiva.
El objetivo primario de ambos estudios de fase III fue evaluar la eficacia de Lyfnua para reducir la frecuencia de tos durante 24 horas en relación con placebo. Los objetivos secundarios fueron la reducción de la frecuencia de la tos en horas de vigilia y la calidad de vida específica de la tos. En ambos estudios, los pacientes fueron aleatorizados para recibir dos veces al día dosis de Lyfnua de
45 mg, 15 mg o placebo. El periodo primario de eficacia en el estudio COUGH-1 (NCT03449134) fue de 12 semanas seguido por un periodo de extensión enmascarado de 40 semanas. El periodo primario de eficacia en el estudio COUGH-2 (NCT03449147) fue de 24 semanas seguido por un periodo de extensión enmascarado de 28 semanas.
Los pacientes incluidos en los estudios COUGH-1 y COUGH-2 no eran fumadores en el momento del reclutamiento, no estaban recibiendo inhibidores de la enzima convertidora de la angiotensina (ECA), habían sido diagnosticados de TCR o TCI y tenían tos crónica desde hacía más de 1 año. La mayoría
de los pacientes eran mujeres (75 %), de raza blanca (80 %) y europeos (53 %) con una media de edad de 58 años (intervalo, 19 a 89) y el 7 % de los pacientes eran mayores de 75 años. Un total de 61,5 % de los pacientes habían sido diagnosticados de TCR, el 38,5 % habían sido diagnosticados de TCI y la media de duración de la tos crónica era de 11 años.
Frecuencia de la tos
En los estudios COUGH-1 y COUGH-2, los pacientes tratados con Lyfnua 45 mg dos veces al día mostraron una reducción significativa de la frecuencia de la tos durante 24 horas en comparación con placebo (Tabla 2). La reducción de la frecuencia de la tos durante 24 horas se observó en la semana 4 y persistió durante todo el periodo primario de eficacia (12 semanas en el estudio COUGH-1 y
24 semanas en el estudio COUGH-2; Figura 1).
El grupo tratado con gefapixant 15 mg dos veces al día no mostró una reducción significativa de la frecuencia de la tos durante 24 horas en ninguno de los estudios.
Tabla 2: resultados de frecuencia de la tos durante 24 horas con Lyfnua 45 mg dos veces al día
(COUGH-1 y COUGH-2)
| COUGH-1 | COUGH-2 | |||
| Lyfnua | Placebo | Lyfnua | Placebo | |
| N | 243 | 243 | 439 | 435 |
| Variable primaria de eficacia | ||||
| Frecuencia de la tos durante 24 horas (toses por hora) | ||||
| Valor inicial (media geométrica) | 18,24 | 22,83 | 18,55 | 19,48 |
| Semana 12 (COUGH-1) o Semana 24 (COUGH-2) (media geométrica) | 7,05 | 10,33 | 6,83 | 8,34 |
| Semana 12 (COUGH-1) o Semana 24 (COUGH-2) (%-reducción respecto al valor inicial) | -61,35 | -54,77 | -63,17 | -57,19 |
| Reducción en relación con placebo (%-reducción e IC del 95 %)† | -18,52 (-32,76; -1,28) | -13,29 (-24,74; -0,10) | ||
| Valor de p | 0,036 | 0,048 | ||
| N = Número de participantes incluidos en el análisis. IC = intervalo de confianza. † Los valores de referencia ausentes se imputaron en función del sexo y la región, seguidos de una imputación múltiple de los datos ausentes (m = 50 conjuntos de datos imputados) para todas las visitas de seguimiento que utilizaban el tratamiento, el sexo, la región y las otras visitas de seguimiento como covariables. Después de la imputación, se realizó un modelo de análisis de covarianza (ANCOVA) en el punto temporal de interés, ajustando las covariables de tratamiento, la situación inicial, el sexo y la región. |
Figura 1: análisis de la frecuencia de la tos durante 24 horas a lo largo del tiempo con Lyfnua
45 mg dos veces al día (COUGH-1 y COUGH-2)
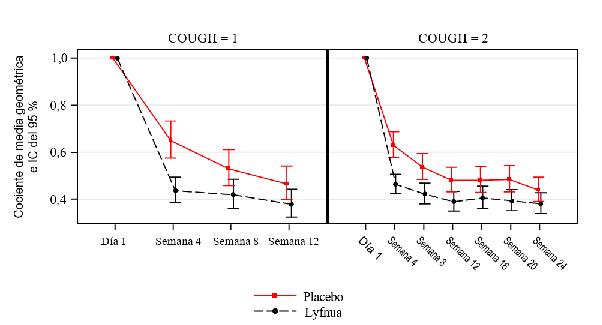
Calidad de vida específica de la tos
El estudio COUGH-2 fue diseñado específicamente para evaluar el impacto de Lyfnua sobre la calidad de vida específica de la tos en relación con placebo, medida mediante el cuestionario de tos de Leicester (LCQ, por sus siglas en inglés) (la puntuación posible va de 3 a 21, de manera que las puntuaciones más altas indican una mejor calidad de vida). Se definió como clínicamente importante
un aumento ≥ 1,3 puntos respecto al valor inicial en la puntuación total del LCQ. En el estudio
COUGH-2, la posibilidad de tener una mejora clínicamente importante de la calidad de vida específica de la tos fue significativamente mayor en el grupo de tratamiento con Lyfnua 45 mg que en el grupo placebo, medido en la semana 24 (ver Tabla 3).
Tabla 3: calidad de vida específica de la tos con Lyfnua 45 mg dos veces al día (COUGH-2):
proporción de pacientes con un aumento ≥ 1,3 puntos con respecto al valor inicial en la puntuación total del LCQ en la semana 24
| Lyfnua | Placebo | |
| N | 439 | 435 |
| Pacientes con respuesta* (%) | 75,7 | 68,1 |
| Cociente de posibilidades estimado frente a placebo (IC del 95 %)† | 1,46 (1,07; 1,99) | |
| Diferencia estimada† frente a placebo (IC del 95 %)†† | 7,63 (1,34; 13,76) | |
| Valor de p† | 0,016 | |
| N = Número de sujetos con datos disponibles en la semana 24. *Porcentaje de pacientes con respuesta en la semana 24. El número de pacientes con respuesta se calculó utilizando un promedio de imputaciones múltiples; hubo aproximadamente 332 y 296 pacientes con respuesta en el grupo de Lyfnua y placebo, respectivamente. IC = intervalo de confianza. LCQ = Leicester Cough Questionnaire (Cuestionario de tos de Leicester). † Los valores de referencia ausentes se imputaron en función del sexo y la región, seguidos de una imputación múltiple de los datos ausentes (m = 50 conjuntos de datos imputados) para todas las visitas de seguimiento que utilizaban el tratamiento, el sexo, la región y las otras visitas de seguimiento como covariables. Después de la imputación, se realizó una regresión logística sobre las puntuaciones dicotómicas en el punto temporal de interés, ajustando las covariables del tratamiento, la puntuación total inicial (continua) del LCQ, el sexo y la región. †† Basado en el método bootstrap. |
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Lyfnua (gefapixant) en todos los grupos de la población pediátrica en el tratamiento de la tos crónica refractaria o idiopática (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
∙
Se estudió la farmacocinética de gefapixant en adultos sanos y en adultos con TCR o TCI y fueron
similares en estas dos poblaciones. El AUC plasmático medio y la concentración máxima (Cmáx) en el estado estacionario son 4.144 ng h/ml y 531 ng/ml con el tratamiento con gefapixant 45 mg dos veces al día. Se alcanza el estado estacionario en el plazo de 2 días, con un cociente de acumulación de 1,4
a 1,5 veces.
Absorción
Después de la administración oral de gefapixant, el tiempo hasta alcanzar las concentraciones plasmáticas máximas (Tmáx) fue de 1 a 4 horas. Los aumentos de la exposición son proporcionales a la dosis después de dosis múltiples de hasta 300 mg dos veces al día. La fracción absorbida de gefapixant es de al menos el 78 %.
Efecto de los alimentos
En relación con las condiciones en ayunas, la administración oral de una dosis única de gefapixant de
50 mg con una comida estándar rica en grasas y rica en calorías no tuvo efecto sobre el AUC o la Cmáx
de gefapixant.
Distribución
De acuerdo con los análisis de farmacocinética poblacional, el volumen de distribución aparente medio en el estado estacionario se calcula que es de 138 l después de la administración oral de una dosis de 45 mg.
In vitro, gefapixant muestra baja unión a proteínas plasmáticas (55 %) y tiene un cociente de sangre a plasma de 1,1. De acuerdo con los estudios preclínicos, gefapixant tiene baja penetración en el SNC.
Biotransformación
El metabolismo hepático es una vía secundaria de eliminación de gefapixant, que conlleva oxidación y glucuronidación. Después de la administración oral de [14C] gefapixant, el 14 % de la dosis administrada se recuperó como metabolitos en la orina y en las heces. El gefapixant inalterado es el principal componente relacionado con el fármaco presente en el plasma (87 %) y cada metabolito circulante representó menos del 10 % de la radiactividad total detectada.
Eliminación
La excreción renal es la vía principal de eliminación de gefapixant y conlleva tanto filtración renal pasiva como mecanismos de transporte activo. Gefapixant se recupera en la orina como compuesto original (~64 %) o metabolitos (~12 %) y el resto se recupera en las heces como compuesto original
(~20 %) o metabolitos (~2 %). Se calcula que la secreción renal activa representa ≤ 50 % de la
eliminación total. In vitro, gefapixant es un sustrato de los transportadores MATE1, MATE2K, glucoproteína-P (gp-P) y proteína de resistencia al cáncer de mama (BCRP, por sus siglas en inglés).
Gefapixant tiene una semivida terminal (t½) de 6 – 10 horas.
Poblaciones especiales
Insuficiencia renal
La excreción renal es la vía principal de eliminación de gefapixant. La insuficiencia renal leve o moderada (TFGe 30 ml/minuto/1,73 m2) no tiene un efecto clínicamente importante sobre la exposición de gefapixant.
En un análisis de farmacocinética poblacional que incluyó a pacientes con tos crónica refractaria o idiopática, se predijo que la media de AUC y Cmáx de gefapixant aumentaban en un 89 % y un 54 %, respectivamente, en pacientes con insuficiencia renal grave (TFGe < 30 ml/minuto/1,73 m2) en comparación con aquellos con función renal normal. Para mantener exposiciones sistémicas similares
a las obtenidas con la función renal normal, se recomienda ajustar la dosis (ver sección 4.2).
Insuficiencia hepática
El metabolismo hepático es una vía secundaria de eliminación. La mayor parte de una dosis oral se recuperó como el compuesto original inalterado en la orina (64 %) o en las heces (20 %). No se realizó un estudio dedicado a sujetos con insuficiencia hepática, porque no es probable que el deterioro de la función hepática tenga un efecto clínicamente importante sobre la exposición (ver sección 4.2).
Efectos de la edad, el peso corporal, el sexo, la etnia y la raza
De acuerdo con un análisis de farmacocinética poblacional, la edad, el peso corporal, el sexo, la etnia y la raza no tienen un efecto clínicamente importante sobre la farmacocinética de gefapixant.
Interacciones medicamentosas
Efectos de otros medicamentos sobre la farmacocinética de gefapixant
El metabolismo hepático es una vía secundaria de eliminación de gefapixant y la posibilidad de interacciones medicamentosas clínicamente importantes de gefapixant con la administración conjunta
de inhibidores o inductores de las enzimas del citocromo P450 (CYP) o uridina ácido 5’-
difosfoglucurónico glucuronosil transferasa (UGT) es baja.
El uso concomitante de un inhibidor de la bomba de protones, omeprazol, no tuvo efecto clínicamente importante sobre la farmacocinética de gefapixant.
De acuerdo con estudios in vitro, gefapixant es un sustrato de los transportadores de expulsión de múltiples fármacos y toxinas 1 (MATE1), MATE2K, glucoproteína-P (gp-P) y proteína de resistencia al cáncer de mama (BCRP). En un estudio clínico de fase 1, una dosis única de pirimetamina, que es un inhibidor de MATE1/MATE2K, aumentó el AUC de gefapixant en un 24 %, una cantidad que no es clínicamente importante y no afectó a la Cmáx de gefapixant.
Efectos de gefapixant sobre la farmacocinética de otros medicamentos
De acuerdo con estudios in vitro, la posibilidad de que gefapixant cause inhibición o inducción de CYP es baja y, por tanto, es poco probable que gefapixant afecte al metabolismo de otros fármacos mediados por CYP.
Gefapixant es un inhibidor de MATE1, MATE2K y del polipéptido transportador de aniones orgánicos 1B1 (OATP1B1) y OATP1B3 in vitro. Sin embargo, el riesgo de interacciones medicamentosas clínicamente importantes mediante la inhibición de estos transportadores es bajo con gefapixant administrado a dosis de 45 mg dos veces al día. No está establecida la importancia clínica de la inhibición in vitro del transportador de cationes orgánicos 1 (OCT1) por gefapixant. En un estudio clínico de fase 1, dosis múltiples de gefapixant 45 mg no afectaron a la exposición de pitavastatina, sustrato de OATP1B.
5.3 Datos preclínicos sobre seguridad
Toxicidad a dosis repetidas
Se produjo cristaluria en animales de laboratorio que recibieron gefapixant y se confirmó que la mayoría de los cristales en orina estaban compuestos de gefapixant.
En un estudio a dosis repetidas durante seis meses en ratas, se observaron cambios microscópicos en el riñón (túbulos dilatados debido a la presencia de material cristalino, degeneración de las células epiteliales que tapizan los túbulos e inflamación del intersticio), en el uréter (dilatación e inflamación) y en la vejiga (hiperplasia de células transicionales) a una exposición 9 veces la exposición en
humanos a la dosis máxima recomendada en humanos (DMRH).
En un estudio de toxicidad oral a dosis repetidas durante nueve meses en perros, se observaron cristales en la orina y la observación microscópica de degeneración tubular focal, mínima, que incluía túbulos corticales ocasionales en un perro macho a una exposición 35 veces mayor que la exposición en humanos a la DMRH.
Carcinogenicidad
Estudios de carcinogenicidad en ratas (de 2 años de duración) y ratones transgénicos rasH2 (de
6 meses de duración) con gefapixant no mostraron pruebas de potencial carcinógeno (no hubo tumores relacionados con el tratamiento) a exposiciones hasta 9 veces (ratas) y 4 veces (ratones) superiores a las exposiciones a la DMRH.
Mutagénesis
Gefapixant no fue genotóxico en una batería de ensayos in vitro o in vivo que incluyeron mutagénesis microbiana, aberración cromosómica en linfocitos de sangre periférica humana y la prueba de micronúcleos de rata in vivo.
Toxicidad para la reproducción
En estudios de reproducción en animales, la administración oral de gefapixant a ratas y conejas gestantes durante el periodo de organogénesis no mostró pruebas de teratogenicidad o letalidad embriofetal a exposiciones (AUC) que fueron 6 veces (ratas) y 34 veces (conejas) la exposición a la DMRH. Se observó una ligera reducción de los pesos fetales en ratas, que se asoció a toxicidad materna, a una exposición aproximadamente 11 veces la exposición a la DMRH.
Los estudios en ratas y conejas gestantes mostraron que gefapixant se transfiere al feto a través de la placenta, con concentraciones plasmáticas fetales hasta el 21 % (ratas) y el 25 % (conejas) de las concentraciones maternas observadas el día 20 de gestación.
En un estudio en la lactancia, gefapixant se excretó en la leche de ratas lactantes cuando se administró por vía oral (hasta 9 veces la exposición a la DMRH) el día 10 de lactancia, con concentraciones en la leche 4 veces la concentración plasmática materna observada 1 hora después de la dosis el día 10 de lactancia.
No hubo efectos sobre la fertilidad, el rendimiento de emparejamiento o el desarrollo embrionario precoz cuando gefapixant se administró a ratas hembra y macho hasta 9 veces la exposición a la DMRH.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido
Sílice coloidal anhidra (E 551) Crospovidona (E 1202) Hipromelosa (E 464)
Estearato de magnesio (E 470b) Manitol (E 421)
Celulosa microcristalina (E 460) Fumarato de estearilo y sodio
Recubrimiento con película
Hipromelosa (E 464) Dióxido de titanio (E 171) Triacetina (E 1518)
Óxido de hierro rojo (E 172) Cera de carnauba (E 903)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Blísteres de PVC/PE/PVdC de color blanco, opacos, con cubierta de lámina de aluminio de abertura a presión.
Envases de 28, 56 y 98 comprimidos recubiertos con película en blísteres no perforados
(14 comprimidos por placa) y envases múltiples con 196 (2 envases de 98) comprimidos recubiertos con película en blísteres no perforados.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme B.V. Waarderweg 39
2031 BN Haarlem
Países Bajos
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/21/1613/001
EU/1/21/1613/002
EU/1/21/1613/003
EU/1/21/1613/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: {DD/mes/AAAA}
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada sobre este medicamento está disponible en la página web de la Agencia
Europea de Medicamentos http://www.ema.europa.eu
ANEXO II
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Merck Sharp & Dohme B.V. Waarderweg 39
2031 BN Haarlem
Países Bajos
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica.
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
· Informes periódicos de seguridad (IPSs)
Los requerimientos para la presentación de los IPSs para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater,
apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
El titular de la autorización de comercialización (TAC) presentará el primer IPS para este medicamento en un plazo de 6 meses después de la autorización.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
· Plan de gestión de riesgos (PGR)
El titular de la autorización de comercialización (TAC) realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el
Módulo 1.8.2 de la autorización de comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
· A petición de la Agencia Europea de Medicamentos.
· Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de
riesgos).
ANEXO III ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
CAJA EXTERIOR

1. NOMBRE DEL MEDICAMENTO
![]()
![]()
Lyfnua 45 mg comprimidos recubiertos con película gefapixant
2. PRINCIPIO(S) ACTIVO(S)
![]()
Cada comprimido recubierto con película contiene 45 mg de gefapixant (como citrato).
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
![]()
![]()
28 comprimidos recubiertos con película
56 comprimidos recubiertos con película
![]()
98 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
![]()
Leer el prospecto antes de utilizar este medicamento. Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
![]()
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
![]()
![]()
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN

10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO, CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
Merck Sharp & Dohme B.V. Waarderweg 39
2031 BN Haarlem
![]()
Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN

![]()
EU/1/21/1613/001 (28 comprimidos recubiertos con película) EU/1/21/1613/002 (56 comprimidos recubiertos con película) EU/1/21/1613/003 (98 comprimidos recubiertos con película)
13. NÚMERO DE LOTE
![]()
Lote
![]()
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
![]()
Lyfnua 45 mg
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
![]()
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC SN NN
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
CAJA EXTERIOR PARA EL ENVASE MÚLTIPLE (CON BLUE BOX)

1. NOMBRE DEL MEDICAMENTO
![]()
![]()
Lyfnua 45 mg comprimidos recubiertos con película gefapixant
2. PRINCIPIO(S) ACTIVO(S)
![]()
Cada comprimido recubierto con película contiene 45 mg de gefapixant (como citrato).
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
![]()
![]()
Envase múltiple: 196 (2 envases de 98) comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
![]()
Leer el prospecto antes de utilizar este medicamento. Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
![]()
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
![]()
![]()
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN

10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO, CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
Merck Sharp & Dohme B.V. Waarderweg 39
2031 BN Haarlem
![]()
Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
EU/1/21/1613/004
13. NÚMERO DE LOTE
![]()
Lote
![]()
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
![]()
![]()
Lyfnua 45 mg
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
![]()
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC SN NN
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
CAJA INTERMEDIA DEL ENVASE MÚLTIPLE (SIN BLUE BOX)

1. NOMBRE DEL MEDICAMENTO
![]()
![]()
Lyfnua 45 mg comprimidos recubiertos con película gefapixant
2. PRINCIPIO(S) ACTIVO(S)
![]()
Cada comprimido recubierto con película contiene 45 mg de gefapixant (como citrato).
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
![]()
![]()
98 comprimidos recubiertos con película. Componente de un envase múltiple, no se puede vender por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
![]()
Leer el prospecto antes de utilizar este medicamento. Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
![]()
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
![]()
![]()
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN

10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO, CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
Merck Sharp & Dohme B.V. Waarderweg 39
2031 BN Haarlem
![]()
Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
EU/1/21/1613/004
13. NÚMERO DE LOTE
![]()
Lote
![]()
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
![]()
![]()
Lyfnua 45 mg
![]()
17. IDENTIFICADOR ÚNICO – CÓDIGO DE BARRAS 2D
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERES O TIRAS
BLÍSTER

1. NOMBRE DEL MEDICAMENTO
![]()
![]()
Lyfnua 45 mg comprimidos gefapixant
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
![]()
MSD
3. FECHA DE CADUCIDAD
![]()
CAD
4. NÚMERO DE LOTE
![]()
Lote
5. OTROS
B. PROSPECTO
Prospecto: información para el paciente
Lyfnua 45 mg comprimidos recubiertos con película
gefapixant
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Lyfnua y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Lyfnua
3. Cómo tomar Lyfnua
4. Posibles efectos adversos
5. Conservación de Lyfnua
6. Contenido del envase e información adicional
1. Qué es Lyfnua y para qué se utiliza
Lyfnua contiene el principio activo gefapixant.
Lyfnua es un medicamento que se utiliza en adultos para la tos crónica (tos que dura más de
8 semanas) y:
· la tos no desaparece incluso después de utilizar otros medicamentos o
· se desconoce la razón de la tos.
El principio activo de Lyfnua, gefapixant, bloquea la acción de los nervios que provocan la tos anormal.
2. Qué necesita saber antes de empezar a tomar Lyfnua
No tome Lyfnua
- sí es alérgico a gefapixant o a alguno de los demás componentes de este medicamento
(incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar y mientras esté tomando Lyfnua si:
- es alérgico a medicamentos que contienen sulfonamida
- tiene apnea del sueño – en la que su respiración se detiene y vuelve a empezar mientras duerme
- desarrolla una infección aguda del pulmón/sistema respiratorio inferior (por ejemplo, neumonía o bronquitis)
- percibe un cambio en el sabor de las cosas, pérdida del gusto o menos capacidad para saborear, que continúa incluso después de dejar de tomar Lyfnua
Niños y adolescentes
No dé este medicamento a niños y adolescentes menores de 18 años de edad. Esto es porque no se ha estudiado en este grupo de edad.
Otros medicamentos y Lyfnua
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento.
Embarazo y lactancia
Se desconoce si Lyfnua pueden dañar al feto. Por lo tanto, es mejor evitar el uso de Lyfnua si está embarazada.
Si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de tomar este medicamento.
Los estudios en animales han demostrado que Lyfnua puede pasar a la leche materna. No se puede descartar un riesgo para su bebé. Usted y su médico deben decidir de forma conjunta si tomar Lyfnua o elegir la lactancia.
Conducción y uso de máquinas
Es posible que se sienta mareado después de tomar Lyfnua. Si esto ocurre, no conduzca ni utilice herramientas o máquinas hasta que deje de sentirse mareado.
Lyfnua contiene sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por comprimido; esto es, esencialmente “exento de sodio”.
3. Cómo tomar Lyfnua
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Qué cantidad tomar
La dosis recomendada de Lyfnua es:
- un comprimido de 45 mg dos veces al día.
Adultos con problemas renales
Su médico puede cambiar la cantidad y la frecuencia con la que toma Lyfnua si:
- tiene insuficiencia renal grave y no está en diálisis.
Como se toma
Trague el comprimido entero. No parta, machaque ni mastique el comprimido. Puede tomar el comprimido con o sin alimentos.
Si toma más Lyfnua del que debe
Si toma demasiado Lyfnua, contacte con un médico o farmacéutico inmediatamente.
Si olvidó tomar Lyfnua
Si olvida una dosis, sáltese esa dosis y tome la siguiente dosis a la hora programada. No tome una dosis doble para compensar las dosis olvidadas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Los posibles efectos adversos son:
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
- cambio en el sabor de las cosas (como sabor a: metálico, amargo o salado)
- tener menos capacidad degustativa
- pérdida del gusto
Frecuentes (pueden afectar a hasta 1 de cada 10 personas)
- sensación de ganas de vomitar (náuseas)
- sabor diferente de las cosas a como sabían antes
- tos (empeoramiento, aumento)
- boca seca
- infección de las vías respiratorias superiores (una infección en la parte superior de las vías respiratorias, que incluye la nariz y la garganta)
- diarrea
- dolor en la boca o la garganta
- tener menos hambre de lo habitual
- sentirse mareado
- dolor en la zona alta del abdomen (vientre)
- indigestión
- sensaciones inusuales en la boca (por ejemplo, hormigueo o sensación de picor)
- pérdida de sensibilidad en la boca
- aumento de la producción de saliva
- insomnio (dificultad para dormir)
Poco frecuentes (pueden afectar a hasta 1 de cada 100 personas)
- piedras en la vejiga, en la orina o en el riñón
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Lyfnua
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el blíster y la caja después de “CAD”. La fecha de caducidad es el último día del mes que se indica.
Este medicamento no requiere condiciones especiales de conservación.
No utilice este medicamento si observa que el envase está dañado o muestra signos de manipulación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Lyfnua
El principio activo es gefapixant. Cada comprimido recubierto con película contiene 45 mg de gefapixant (como citrato).
Los demás componentes son sílice (coloidal anhidra) (E 551), crospovidona (E 1202), hipromelosa (E 464), estearato de magnesio (E 470b), manitol (E 421), celulosa microcristalina (E 460), fumarato de estearilo y sodio. Los comprimidos están recubiertos con película con un material de recubrimiento que contiene los siguientes componentes: hipromelosa (E 464), dióxido de titanio (E 171), triacetina
(E 1518) y óxido de hierro rojo (E 172). Los comprimidos están pulidos con cera de carnauba (E 903).
Aspecto del producto y contenido del envase
Lyfnua es un comprimido de color rosa, redondo y convexo, grabado con “777” en una cara y liso en la otra.
Lyfnua está disponible en blísteres blancos de PVC/PE/PVdC.
Lyfnua está disponible en envases de 28, 56 y 98 comprimidos recubiertos con película en blísteres no perforados (14 comprimidos por placa) y envases múltiples con 196 (2 envases de 98) comprimidos recubiertos con película en blísteres no perforados.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Merck Sharp & Dohme B.V. Waarderweg 39
2031 BN Haarlem
Países Bajos
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
België/Belgique/Belgien
MSD Belgium
Tél/Tel: +32(0)27766211 [email protected]
Lietuva
UAB Merck Sharp & Dohme
Tel. + 370 5 278 02 47 [email protected]
България
Мерк Шарп и Доум България ЕООД Тел.: +359 2 819 3737
Luxembourg/Luxemburg
MSD Belgium
Tél/Tel: +32(0)27766211 [email protected]
Česká republika
Merck Sharp & Dohme s.r.o. Tel: +420 233 010 111 [email protected]
Magyarország
MSD Pharma Hungary Kft. Tel.: +36 1 888 5300 [email protected]
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 [email protected]
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) [email protected]
Deutschland
MSD Sharp & Dohme GmbH
Tel: 0800 673 673 673 (+49 (0) 89 4561 0)
[email protected]
Nederland
Merck Sharp & Dohme B.V. Tel: 0800 9999000
(+31 23 5153153)
[email protected]
Eesti
Merck Sharp & Dohme OÜ Tel.: +372 6144 200
[email protected]
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00
[email protected]
Ελλάδα
MSD Α.Φ.Β.Ε.Ε.
Τηλ: +30 210 98 97 300 [email protected]
Österreich
Merck Sharp & Dohme Ges.m.b.H. Tel: +43 (0) 1 26 044
[email protected]
España
Merck Sharp & Dohme de España, S.A. Tel: +34 91 321 06 00 [email protected]
Polska
MSD Polska Sp. z o.o. Tel: +48 22 549 51 00 [email protected]
France
MSD France
Tél: + 33 (0) 1 80 46 40 40
Portugal
Merck Sharp & Dohme, Lda
Tel: +351 21 4465700 [email protected]
Hrvatska
Merck Sharp & Dohme d.o.o. Tel: + 385 1 6611 333
[email protected]
România
Merck Sharp & Dohme Romania S.R.L. Tel: +40 21 529 29 00
[email protected]
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700
[email protected]
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel: +386 1 5204 201
[email protected]
Ísland
Vistor hf.
Sími: + 354 535 7000
Slovenská republika
Merck Sharp & Dohme, s. r. o. Tel: +421 2 58282010
[email protected]
Italia
MSD Italia S.r.l.
Tel: 800 23 99 89 (+39 06 361911)
[email protected]
Suomi/Finland
MSD Finland Oy
Puh/Tel: +358 (0)9 804 650 [email protected]
Κύπρος
Merck Sharp & Dohme Cyprus Limited
Τηλ.: 800 00 673 (+357 22866700)
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 [email protected]
Latvija
SIA Merck Sharp & Dohme Latvija Tel: + 371 67364224 [email protected]
United Kingdom (Northern Ireland)
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700
[email protected]
Fecha de la última revisión de este prospecto: {MM/AAAA}.
La información detallada de este medicamento está disponible en la página web de la Agencia
Europea de Medicamentos: http://www.ema.europa.eu

