Para recomendaciones de dosificación en diferentes grupos de edad, ver sección 13. Dosis y Vía de Administración.
Farmacocinética:
El levetiracetam es un compuesto altamente soluble y permeable. Su perfil farmacocinético es lineal e independiente del tiempo con baja variabilidad intra e intersujetos. No se presenta modificación de la depuración después de la administración de dosis repetidas. Tampoco hay evidencias relevantes de variabilidad relacionadas con el género, la raza o el ritmo circadiano. El perfil farmacocinético es comparable en voluntarios sanos y en pacientes con epilepsia.
Debido a su absorción completa y lineal, pueden predecirse los niveles plasmáticos en mg/kg de peso, a partir de una dosis de levetiracetam administrada por vía oral. Por lo que no es necesario monitorear los niveles plasmáticos de levetiracetam.
En niños y adultos se ha observado una correlación significativa entre las concentraciones plasmáticas y en saliva de levetiracetam (la relación de concentración saliva/plasma se encuentra en el rango de 1 a 1.7 para las tabletas orales y 4 horas después de administrar la solución oral).
Adultos y adolescentes
Absorción
La biodisponibilidad de las tabletas de liberación prolongada es similar a la de las tabletas de liberación inmediata. La farmacocinética (ABC y Cmáx) mostró ser proporcional después de la administración de la dosis única de 1,000 mg, 2,000 mg y 3,000 mg de tabletas de liberación prolongada de levetiracetam. Las concentraciones plasmáticas pico (Cmáx) se alcanzan en alrededor de 4 horas. La administración individual de dos tabletas de liberación prolongada de 500 mg una vez al día produjo concentraciones plasmáticas máximas comparables, y de área bajo la concentración plasmática frente al tiempo, como sucedió con la administración de una tableta de liberación inmediata de 500 mg dos veces al día en ayunas. Después de múltiples dosis con tabletas de liberación prolongada, la tasa de exposición (ABC0-24) fue similar a la tasa de exposición después de múltiples dosis con tabletas de liberación inmediata. Cmáx y Cmin fueron más bajas por 17% y 26% respectivamente, después de dosis de tabletas de liberación prolongada en comparación con tabletas de liberación inmediata a dosis múltiples.
Dos tabletas de liberación prolongada de 750 mg fueron bioequivalentes a una única administración de tres tabletas de liberación prolongada de 500 mg.
La administración de tabletas de liberación prolongada en estado postprandial resultó en una concentración pico mayor y en un tiempo medio a pico mayor (Tmáx de 2 horas más).
La relación entre la efectividad de la misma dosis diaria de tabletas de liberación prolongada y tabletas de liberación inmediata no ha sido estudiada y es desconocida.
Levetiracetam es liberado lentamente mediante difusión del gel que se forma después de la deglución (cuando la matriz de hipromelosa de la tableta está en contacto con el agua o con los líquidos fisiológicos) y es absorbido a lo largo del tracto intestinal.
Los pacientes podrán ocasionalmente observar en las heces la matriz de la tableta vacía.
Distribución
No se cuenta con datos disponibles de distribución en tejido humano. Ni el levetiracetam ni su metabolito primario se unen significativamente a las proteínas plasmáticas (< 10%). El volumen de distribución del levetiracetam es de aproximadamente 0.5 a 0.7 l/kg, un valor cercano al volumen corporal total de agua.
Biotransformación
El levetiracetam no es metabolizado extensamente en humanos. Su vía metabólica principal (24% de la dosis) es la hidrólisis enzimática del grupo acetamida. La producción del metabolito primario, ucb L057, no es mediada por las isoformas del citocromo P450 hepático. La hidrólisis del grupo acetamida fue cuantificable en gran número de tejidos, incluyendo a las células sanguíneas. El metabolito ucb L057 es farmacológicamente inactivo.
También se identificaron 2 metabolitos menores. Uno, obtenido por la hidroxilación del anillo pirrolidona (1.6% de la dosis) y el otro, por la apertura del anillo pirrolidona (0.9% de la dosis).
Otros compuestos no identificados participaron únicamente con el 0.6% de la dosis.
No se evidenció interconversión enantiomérica in vivo para el levetiracetam o para su metabolito principal.
En ensayos in vitro, el levetiracetam y su metabolito primario mostraron que no inhiben la actividad de las isoformas hepáticas principales del citocromo P450 (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 y 1A2) en humanos, de la glucuronil transferasa (UGT1A1 y UGT1A6) y de la actividad hidroxilasa epóxica. Además, el levetiracetam no afecta la glucuronización in vitro del ácido valproico.
En cultivo de hepatocitos humanos, el levetiracetam tiene un efecto mínimo o nulo en la conjugación del etinilestradiol o de CYP1A1/2. Levetiracetam causa una inducción moderada de CYP2B6 y CYP3A4 a concentraciones elevadas (680 μg/mL), sin embargo, a concentraciones aproximadas a Cmáx, que se alcanza después de aplicaciones repetidas de 1,500 mg/dos veces/día, los efectos no fueron considerados biológicamente relevantes. Debido a todo lo anterior, la interacción de Keppra® XR con otras sustancias o viceversa es muy improbable.
Eliminación
La vida media plasmática en adultos es de 7 ± 1 horas y no varía con la dosis, vía de administración o administración repetida. El promedio de la depuración corporal total es de 0.96 ml/min/kg.
La principal vía de eliminación es la urinaria, con por lo menos el 95% de la dosis, (aproximadamente el 93% de la dosis se excretó en un periodo de 48 hrs.). La excreción por vía fecal representa únicamente el 0.3% de la dosis.
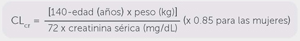
La excreción urinaria acumulativa del levetiracetam y su metabolito primario durante las primeras 48 horas, registró el 66% y el 24% de la dosis. La depuración renal de levetiracetam y de ucb L057 es de 0.6 y 4.2 ml/min/kg respectivamente, lo que indica que el levetiracetam es excretado por filtración glomerular con la subsecuente reabsorción tubular y que el metabolito primario también se excreta por secreción tubular activa además de la filtración glomerular. La eliminación de levetiracetam se correlaciona con la depuración de creatinina.
Ancianos
En los ancianos, la vida media se incrementa en aproximadamente un 40% (10 a 11 h). Esto se relaciona con la disminución de la función renal en esta población.
Insuficiencia renal
La depuración corporal aparente está correlacionada con la depuración de creatinina, tanto para levetiracetam como para su metabolito primario. Por lo que se recomienda ajustar la dosis diaria de mantenimiento de Keppra® XR, basándose en la depuración de creatinina de los pacientes con insuficiencia renal moderada y severa.
En pacientes adultos con patología renal terminal anúrica, la vida media fue aproximadamente de 25 y de 3.1 horas durante los períodos interdiálisis e intradiálisis respectivamente La fracción de levetiracetam eliminada durante una sesión de diálisis normal de 4horas fue de un 51%.
Insuficiencia hepática
No se presentó modificación relevante de la depuración de levetiracetam en los sujetos con insuficiencia hepática leve (Child-Pugh A) a moderada (Child-Pugh B), la farmacocinética del levetiracetam resulto sin cambios. En sujetos con insuficiencia hepática severa (Child-Pugh C), la depuración total fue del 50% de la de sujetos normales, pero la depuración renal disminuida explica la mayor parte de la reducción.
No se necesita ajustar la dosis en pacientes con insuficiencia hepática leve a moderada. En pacientes con insuficiencia hepática severa, la depuración de creatinina puede subestimar la insuficiencia renal. Por lo tanto, se recomienda una reducción del 50% de la dosis diaria de mantenimiento cuando la depuración de creatinina es < 60 ml/min/1.73m2.
Niños (12 a 16 años)
Se realizó un estudio de etiqueta abierta para comparar la farmacocinética de las tabletas de liberación prolongada en niños de 12 a 16 años de edad, con la farmacocinética en pacientes adultos posterior a la administración repetida. El ABC y la Cmáx fueron equivalentes en los dos grupos de edad.
Farmacodinamia:
Grupo Farmacoterapéutico
Antiepiléptico, código ATC: N03AX14.
La sustancia activa, levetiracetam, es un derivado de la pirrolidona (S-enantiómero de α-etil-2-oxo-1-pirrolidinacetamida), no relacionado químicamente con las sustancias activas antiepilépticas existentes.
Mecanismo de acción
El mecanismo de acción del levetiracetam no ha sido totalmente dilucidado, pero parece ser diferente a los mecanismos de los fármacos antiepilépticos actuales. Los estudios in vivo e in vitro sugieren que el levetiracetam no altera las características celulares básicas y la neurotransmisión normal.
Los estudios in vitro muestran que el levetiracetam afecta los niveles de Ca2+ intraneuronal por inhibición parcial de las corrientes de Ca2+ de tipo N y por reducción de la liberación de Ca2+ de las reservas intraneuronales. Adicionalmente, revierte parcialmente las reducciones en las corrientes generadas por GABA y glicina inducidas por zinc y β-carbolinas. Más aún, el levetiracetam ha mostrado en estudios in vitro que se une a sitios específicos del tejido cerebral de los roedores. Este sitio de unión es la proteína de la vesícula sináptica 2A, que se cree está involucrada en la fusión de las vesículas y liberación de neurotransmisores. El levetiracetam y los análogos relacionados muestran un rango de afinidad para la unión de la proteína de la vesícula sináptica 2A que correlaciona con la potencia de su protección anticonvulsiva en el modelo audiogénico de epilepsia del ratón. Este hallazgo sugiere que la interacción entre levetiracetam y la proteína de la vesícula sináptica 2A parece contribuir al mecanismo de acción antiepiléptico del fármaco.
Efectos farmacodinámicos
El levetiracetam induce protección contra las crisis en un amplio rango de modelos animales de crisis generalizadas primarias y parciales (focales) sin tener efecto pro-convulsivo. Su metabolito principal es inactivo.
En el hombre, la actividad en las condiciones de epilepsias parciales (focales) y generalizadas (descargas epileptiformes/ respuestas fotoparoxísticas) han confirmado el amplio espectro de su perfil farmacológico preclínico.
Estudios Clínicos de Eficacia con la Formulación de Liberación Prolongada
Monoterapia
La eficacia de las tabletas de liberación prolongada de levetiracetam en la conversión a monoterapia se estableció en un estudio de asignación aleatoria , doble ciego, multicéntrico, de control histórico con un periodo de evaluación de 18 semanas (esto es, un periodo de valoración de 2 semanas, un periodo de retiro de 6 semanas para los medicamentos antiepilépticos precedentes, seguidas de un periodo de monoterapia de 10 semanas) en pacientes de 12 a 75 años de edad con crisis de inicio parcial (focal). En dicho estudio, los pacientes estaban recibiendo dosis estables de un máximo de 2 medicamentos antiepilépticos durante el período basal de 8 semanas. Los pacientes que estaban tomando 2 medicamentos antiepilépticos debían estar tomando < 50% de la dosis de mantenimiento mínima recomendada para 1 de los 2 medicamentos antiepilépticos. Los pacientes se asignaron aleatoriamente para recibir en última instancia ya fuera levetiracetam 1000 mg/día ó 2000 mg/día, y se compararon sus resultados con el grupo de control histórico. Se consideró que la superioridad estadística sobre el control histórico quedaría demostrada si el límite superior de un intervalo de confianza bilateral al 95% para el porcentaje de pacientes que cumplieran el criterio de salida y a quienes se administró levetiracetam, permanecía por debajo del límite inferior de predicción del 95% siendo de 67.8% derivado de los datos del control histórico.
El paciente se retiraba del estudio si cumplía uno o más de los siguientes criterios: (1) un incremento al doble de la frecuencia de crisis parciales (focales) en cualquier momento del periodo de tratamiento de 4 semanas en comparación a la línea basal, (2) un incremento del doble de la frecuencia más elevada de crisis en 2 días consecutivos ocurridas durante el período basal, (3) Que ocurra una crisis generalizada si no se había presentado ninguna en los 6 meses previos a la asignación aleatoria, (4) un episodio de estatus epiléptico o una prolongación o empeoramiento en la duración o frecuencia de la crisis, que a juicio del investigador requiera intervención.
Para el grupo con 2000 mg/día de levetiracetam, la estimación del porcentaje de los pacientes que cumplían con al menos 1 criterio de salida fue del 37.5% (IC 95%: 29.7%, 45.3%). El límite superior del IC 95% bilateral (45.3%) estuvo por debajo del límite inferior del 67.8% derivado de los datos del control histórico.
Terapia Adjunta
La eficacia de las tabletas de levetiracetam de liberación prolongada como terapia concomitante (en adición a otros medicamentos antiepilépticos) fue establecida en un estudio clínico multicéntrico, aleatorizado, controlado con placebo, a través de 7 países en pacientes que tenían crisis refractarias de inicio parcial (focal) con o sin generalización secundaria. Los pacientes enrolados tuvieron al menos ocho crisis parciales (focales) con o sin generalización secundaria durante el periodo basal de 8 semanas y al menos dos crisis parciales (focales) en cada intervalo de 4 semanas del periodo basal. Los pacientes estaban tomando un régimen de dosis estable de al menos uno, y podían tomar un máximo de tres FAEs. Después de un periodo basal de 8 semanas, se aleatorizaron a 158 pacientes para recibir placebo (N=79) o tabletas de levetiracetam de liberación prolongada (2 tabletas de 500 mg) (N=79) administradas una vez al día durante un periodo de tratamiento de 12 semanas.
El criterio de evaluación primario de eficacia fue la frecuencia semanal de crisis de inicio parcial (focal) (Tipo I) durante el periodo de tratamiento. El porcentaje de reducción versus el placebo en la frecuencia semanal de crisis de inicio parcial (focal) durante el periodo de tratamiento fue de 14.4% (estadísticamente significativa; LEV n = 75; PBO n = 78).