Farmacocinética:
Las tabletas, solución oral y solución para inyección intravenosa son bioequivalentes.
Brivaracetam exhibe una farmacocinética lineal e independiente de tiempo con baja variabilidad intra y entre sujetos, asimismo presenta una absorción completa, muy baja unión a proteínas, excreción renal después de una biotransformación extensiva, y metabolitos inactivos a nivel farmacológico.
Brivaracetam tiene un bajo potencial de interacción entre fármacos.
La farmacocinética de brivaracetam es similar cuando se usa como monoterapia o como terapia de adición para el tratamiento de las crisis de inicio parcial.
Absorción
Brivaracetam se absorbe rápida y completamente después de la administración oral. La farmacocinética es proporcional a la dosis a partir de 10 a 600 mg. El Tmáx promedio para las tabletas administradas sin alimentos es de una hora (el rango del Tmáx es de 0.25 a 3 horas).
La administración conjunta con alimentos altos en grasa retarda la tasa de absorción del medicamento mientras que el alcance de absorción permaneció sin cambios.
Distribución
Brivaracetam se une débilmente (≤ 20%) a las proteínas plasmáticas. El volumen de distribución es de 0.5 L/kg, un valor cercano al del total de agua en el organismo.
Debido a su favorable lipofilicidad (Log P), que resulta en la alta permeabilidad de la membrana celular, el brivaracetam penetra rápidamente en el cerebro. Este medicamento se distribuye rápida y uniformemente en la mayoría de los tejidos. En roedores, la proporción de la concentración cerebro a plasma se equilibra con rapidez, indicando la rápida penetración en el cerebro y se acerca a 1, indicando la ausencia del transporte activo.
Metabolismo
Brivaracetam se metaboliza principalmente por medio de hidrólisis de la mitad de amida para formar el ácido carboxílico correspondiente, y posteriormente a través de la hidroxilación en la cadena lateral de propilo. La hidrólisis de la mitad de amida que lleva al metabolito del ácido carboxílico (34% de la dosis en orina), se apoya por la amidasa hepática y extrahepática (E.C.3.5.1.4). In vitro, la hidroxilación del brivaracetam se regula básicamente por medio del CYP2C19. In vivo, en sujetos humanos que tienen mutaciones inefectivas del CYP2C19, la producción del metabolito hidroxi se disminuye en 10 veces mientras que el medicamento citado por sí mismo se incrementa en 22% ó 42% en individuos con uno o ambos alelos mutados. Por lo tanto, no es probable que los inhibidores del CYP2C19 tengan un efecto significativo sobre el brivaracetam. Un metabolito adicional (el metabolito ácido hidroxi) se crea principalmente por hidroxilación de la cadena lateral de propilo en el metabolito ácido carboxílico (principalmente por CYP2C9). Los tres metabolitos son inactivos a nivel farmacológico.
Eliminación
Brivaracetam se elimina principalmente por biotransformación y por excreción en orina. Más del 95% de la dosis, incluyendo metabolitos, se excreta en la orina dentro de las 72 horas después de su toma. Menos del 1% de brivaracetam se excreta en heces y menos del 10% se excreta sin cambios en la orina. La vida media terminal del plasma (t1/2) es aproximadamente de nueve horas.
Poblaciones especiales
- Ancianos
En un estudio en personas de edad avanzada (65 a 79 años de edad; con depuración de creatinina de 53 a 98 ml/min/1.73 ml2), que reciben 400 mg/día de brivaracetam en administración bid, la vida media de brivaracetam en plasma fue de 7.9 y 9.3 horas en grupos de 65 a 75 y > 75 años, respectivamente. La depuración en plasma en estado permanente de brivaracetam fue ligeramente inferior (0.76 ml/min/kg), que en sujetos masculinos saludables jóvenes (0.83 ml/min/kg). No se requiere ningún ajuste de dosis.
- Insuficiencia renal
Un estudio sobre sujetos con insuficiencia renal grave (depuración de creatinina de < 30 ml/min/1.73 m2 y que no requirieron de diálisis), reveló que el ABC de plasma de brivaracetam fue moderadamente incrementada (+ 21%) en relación con controles sanos, mientras que la ABC del ácido, hidroxi y metabolitos de ácido-hidroxi se incrementaron en 3-, 4- y 21- veces, respectivamente. La depuración renal de estos metabolitos no activos disminuyó 10 veces. El metabolito del ácido-hidroxi no reveló ninguna inquietud de seguridad en estudios no clínicos. No se requiere ningún ajuste de dosis para pacientes con insuficiencia leve, moderada o grave. Brivaracetam no ha sido estudiado en pacientes que se tratan con hemodiálisis.
- Insuficiencia hepática
Un estudio farmacocinético sobre sujetos con cirrosis hepática (Child-Pugh, grados A, B y C), mostró incrementos similares en la exposición a brivaracetam independientemente de la gravedad de la enfermedad (50%, 57% y 59%), en relación con los controles sanos comparados. Los ajustes de dosis se recomiendan para pacientes que sufren de insuficiencia hepática. Se espera que el efecto de la insuficiencia hepática en la farmacocinética de brivaracetam en pacientes pediátricos sea comparable al efecto observado en adultos.
- Población pediátrica
Se realizó un estudio farmacocinético abierto, de brazo único, multicéntrico con un período de evaluación de 3 semanas y un ajuste fijo de 3 pasos con Briviact® solución oral en 59 pacientes pediátricos de 4 años y menores de 16 años. En esos pacientes, las concentraciones plasmáticas fueron proporcionales a la dosis. Fue necesario un régimen de dosificación basado en el peso corporal para lograr que las exposiciones de brivaracetam en pacientes pediátricos de 4 años a menores de 16 años fuesen similares a las observadas en adultos tratados con dosis efectivas de Briviact®. El aclaramiento plasmático estimado fue de 1.61 L/h; 2.18 L/h; 3.19 L/h en los pacientes pediátricos que pesaban 20 Kg, 30 Kg y 50 Kg, respectivamente. En comparación, el aclaramiento plasmático se estimó en 3.58 L/h en pacientes adultos (70 Kg de peso corporal).
- Género
No se reportan diferencias en la farmacocinética del brivaracetam por género.
- Raza
La farmacocinética del brivaracetam no se vio afectada significativamente por la raza (caucásica, negra / afroamericana, asiática, amerindia, nativos de Alaska, hispana / latina), en un modelado de la farmacocinética de la población de los pacientes que sufren epilepsia.
Farmacodinamia:
Grupo farmacoterapéutico:
Antiepiléptico, otros antiepilépticos, código ATC: N03AX23.
La sustancia activa es (2S)-2-[(4R)-2-oxo-4-propiltetrahidro-1H-pirrol-1-ilo] butanamida.
Mecanismo de acción
Brivaracetam despliega una afinidad alta y selectiva por la proteína de la vesícula sináptica 2A (SV2A) en el cerebro. Se cree que la unión a la SV2A es el mecanismo primario de la actividad antiepiléptica del brivaracetam.
Los datos farmacodinámicos preclínicos de brivaracetam sugieren una alta potencia y eficacia en la supresión de crisis en diversos tipos de epilepsia en animales incluyendo crisis parciales y generalizadas, así como contra el estado epiléptico y mioclónico.
Interacciones con el alcohol
En un estudio sobre la interacción en sujetos sanos los efectos de brivaracetam sobre las funciones psicomotoras, atención y memoria incrementaron las consecuencias de la discapacidad del alcohol.
Abuso potencial
Un estudio realizado con el brivaracetam no mostró un potencial relacionado con el abuso, pero este potencial todavía no puede excluirse por completo.
Dependencia
No existe evidencia de dependencia potencial física o de síndrome de descontinuación con el brivaracetam en una revisión combinada de estudios de terapia adjunta controlada con placebo.
Efectos en el intervalo QT
El efecto de brivaracetam en la prolongación de QTc se evaluó en un estudio de grupo paralelo controlado por placebo y de doble ciego de selección aleatoria de control positivo (400 mg de moxifloxacino) de brivaracetam (150 y 800 mg/día en dos tomas diarias) en 184 sujetos saludables. No se reportó evidencia de que brivaracetam prolongue el intervalo QT.
Frecuencia de las crisis epilépticas
Una correlación significativa a nivel estadístico ha sido demostrada entre la concentración de brivaracetam en plasma y la reducción de la frecuencia de las crisis epilépticas desde el inicio en los estudios clínicos de confirmación en el tratamiento adjunto de las crisis de inicio parcial. Se estimó que el EC50 (concentración de brivaracetam en plasma correspondiente al 50% del efecto máximo) era de 0.57 mg/L. Esta concentración de plasma está ligeramente arriba de la exposición promedio obtenida después de las dosis del medicamento mencionado de 50 mg/día. Posteriormente la reducción de la frecuencia de crisis epilépticas se obtiene por el incremento de la dosis a 100 mg/día y alcanza una meseta en 200 mg/día.
Estudios clínicos.
La eficacia de brivaracetam como terapia adjunta de la crisis de inicio parcial fue establecida en tres estudios clínicos multicéntricos, de dosis fija, aleatorizados, placebo controlados, doble ciego en sujetos de 16 años de edad y mayores.
La dosis diaria del brivaracetam varió de 5 a 200 mg/día a través de estos estudios. Todos los estudios tuvieron un período inicial de ocho semanas seguido por un período de tratamiento de 12 semanas sin titulación ascendente. Un número de 1,558 pacientes recibió el fármaco del estudio de los cuales, 1,099 recibieron brivaracetam. El criterio de reclutamiento de los estudios requirió que los pacientes tuvieran crisis de inicio parcial no controlado con uno o dos antiepilépticos concomitantes. Se requirió que los pacientes tuvieran, por lo menos ocho crisis de inicio parcial durante el período inicial.
Los antiepilépticos tomados con mayor frecuencia en el período del registro del estudio fueron carbamazepina (40.6%), lamotrigina (25.2%), valproato (20.5%), oxcarbazepina (16.0%), topiramato (13.5%), fenitoína (10.2%) y levetiracetam (9.8%). En el estudio N01358, el 18.9% de los sujetos tenía un historial de 0-1 antiepiléptico previo, un 33.8% de 2 - 4 antiepilépticos previos, y el 47.2% ≥ 5 antiepilépticos previos. La frecuencia promedio inicial de crisis a través de los tres estudios fue de 9 por 28 días. Los pacientes habían tenido una duración promedio de epilepsia de aproximadamente 23 años.
Las valoraciones de la eficacia se resumen en la Tabla 2. En general, el brivaracetam fue eficaz para el tratamiento adjunto de las crisis de inicio parcial con o sin generalización secundaria en pacientes de 16 años de edad y mayores, entre 50 y 200 mg/día.
Los resultados del porcentaje de reducción sobre el placebo del N01252 y del N01253 se basan en la frecuencia de las crisis de inicio parcial por 28 días para permitir la comparación de los resultados presentados en el N01358, aunque los análisis sobre la eficacia primaria del N01252 y del N01253 se basaron en la frecuencia de crisis de inicio parcial por siete días. Las conclusiones respecto a la importancia estadística no se impactaron por el cambio de la duración en la que la frecuencia de la crisis de inicio parcial se estandarizó para los estudios N01252 y N01253.
Tabla 2: Valoraciones Clave de la Eficacia respecto a la Frecuencia de crisis de inicio parcial por 28 días.
| Estudio | Placebo | Brivaracetam
* Significativos a nivel estadístico (valor p) |
50
mg/día | 100
mg/día | 200
mg/día |
| Estudio N01253(1) | | | | |
| n = 96 | n = 101 | | |
| Porcentaje de reducción sobre el placebo (%) | NA | 22.0*
(p=0.004) | ~ | ~ |
| Tasa de respuesta**(%) | 16.7 | 32.7*
(p=0.008) | ~ | ~ |
| Porcentaje de reducción promedio desde el inicio (%) | 17.8 | 30.5*
(p=0.003) | ~ | ~ |
| Estudio N01252(1) | | | | |
| n = 100 | n = 99 | n = 100 | |
| Porcentaje de reducción sobre el placebo (%) | NA | 9.2
(p=0.274) | 20.5(2)
(p=0.010) | ~ |
| Tasa de respuesta** (%) | 20.0 | 27.3
(p=0.372) | 36.0(2)
(p=0.023) | ~ |
| Porcentaje de reducción promedio desde el inicio (%) | 17.0 | 26.8
(p=0.092) | 32.5(2)
(p=0.004) | ~ |
| Estudio N01358 | | | | |
| n = 259 | | n = 252 | n = 249 |
| Porcentaje de reducción sobre el placebo (%) | NA | ~ | 22.8*
(p<0.001) | 23.2*
(p<0.001) |
| Tasa de respuesta** (%) | 21.6 | ~ | 38.9*
(p<0.001) | 37.8*
(p<0.001) |
| Porcentaje de reducción promedio desde el inicio (%) | 17.6 | ~ | 37.2*
(p<0.001) | 35.6*
(p<0.001) |
n=pacientes seleccionados de forma aleatoria que recibieron, por lo menos, una dosis del medicamento del estudio.
~Dosis no estudiada.
*Significativo a nivel estadístico
**Tasa de Respuesta: definida como porcentaje de pacientes que lograron, por lo menos, un 50% de reducción en la frecuencia de la crisis de inicio parcial por 28 días a partir del inicio hasta el período de tratamiento.
(1) Aproximadamente el 20% de los pacientes estuvo tomando levetiracetam concomitante.
(2) La valoración primaria del N01252 no logró una significancia estadística con base en el procedimiento de prueba secuencial, lo que requirió de importancia estadística en el nivel de 0.050 de 50 mg/día de brivaracetam versus placebo previo a la prueba de 100 mg/día de brivaracetam. La dosis de 100 mg/día fue nominalmente significativa.
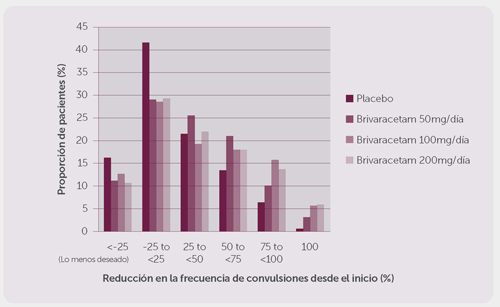
La figura 1 muestra el porcentaje de pacientes (excluyendo los pacientes con levetiracetam concomitante) por categoría de reducción desde el inicio en la frecuencia de la crisis de inicio parcial por 28 días a través de los tres estudios. Los pacientes con más de un 25% de incremento en el padecimiento mencionado se muestran a la izquierda de “lo menos deseable”. Los pacientes con una mejoría en la reducción del procentaje desde el inicio en la frecuencia de dicho padecimiento se presentan en cuatro categorías a la derecha. Los porcentajes de pacientes con, al menos, un 50% de reducción en la frecuencia de las crisis fueron de 20.3%, 34.2%, 39.5% y 37.8% para el placebo, 50 mg/día, 100 mg/día, y 200 mg/día, respectivamente.
Figura 1
La conclusión de que brivaracetam es efectivo como monoterapia en pacientes de 4 años y mayores se extrapoló a partir de los estudios controlados en epilepsia concomitante. Las simulaciones farmacocinéticas y farmacodinámicas mostraron que la dosis de brivaracetam, al ser utilizado como monoterapia, da como resultado una exposición y una exposición-respuesta que son similares a aquellas que han demostrado ser seguras y efectivas cuando se utiliza como terapia de adición para el tratamiento de las crisis de inicio parcial.
Tratamiento con levetiracetam
En los estudios N01252 y N01253, el levetiracetam se administró como antiepiléptico concomitante en aproximadamente el 20% de los pacientes. En el tercer estudio, N01358, el levetiracetam no se permitió como antiepiléptico concomitante.
Aunque el número de pacientes es limitado, no se observó beneficio del brivaracetam contra el placebo en pacientes que toman levetiracetam de manera concurrente. No se observaron problemas de seguridad ni de tolerabilidad adicionales.
En el estudio N01358, un análisis previamente especificado muestra que en pacientes con exposición previa al levetiracetam, se demostró la eficacia sobre el placebo en relación con dosis de 100 y 200 mg/día.
Poblaciones especiales
- Población de edad avanzada
Los tres estudios pivotales controlados con placebo doble ciego incluyeron 38 pacientes de edad avanzada entre 65 y 80 años. Aunque los datos son limitados, la eficacia fue comparable con pacientes más jóvenes. - Población pediátrica
Se ha establecido la seguridad y eficacia de las tabletas de Briviact® y la solución oral en pacientes pediátricos de 4 años a menores de 16 años. El uso de Briviact® en estos grupos de edad está respaldado por la evidencia de estudios adecuados y bien controlados de Briviact® en adultos con crisis de inicio parcial, datos farmacocinéticos de pacientes adultos y pediátricos y datos de seguridad en 149 pacientes pediátricos de 4 años a menos de 16 años de edad.
No se ha establecido la seguridad de la inyección de Briviact® en pacientes pediátricos.
No se han establecido la eficacia y tolerabilidad de brivaracetam en pacientes menores de 4 años de edad.
Estudios de extensión de etiqueta abierta
En todos los estudios, el 81.7% de los pacientes que completaron los estudios pivotales aleatorizados fueron registrados en los estudios de extensión de etiqueta abierta. Desde el ingreso a estudios de asignación aleatoria el 5.3% de los pacientes expuestos al brivaracetam durante seis meses (n = 1500) estuvo libre de crisis en comparación con el 4.6% y el 3.7% de los pacientes expuestos durante 12 meses (n = 1188) y 24 meses (n = 847), respectivamente.