 BEYFORTUS 100 MG SOLUCION INYECTABLE EN JERINGA PRECARGADA
BEYFORTUS 100 MG SOLUCION INYECTABLE EN JERINGA PRECARGADA

| ATC: Nirsevimab |
| PA: Nirsevimab |
Envases
- Env. con 1 jeringa precargada de 1 ml
- H: Medicamento de uso hospitalario
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica- No sustit.: Medicamento NO sustituible por el farmacéutico (Biológicos)
- Fi: Medicamento financiado sólo para determinadas indicaciones
- Facturable SNS: NO
- Comercializado: Si
- Situación: Alta
- Código Nacional: 762405
- EAN13: 8470007624059
- Conservar en frío: Sí
-
PROBLEMAS DE SUMINISTRO
Fecha prevista de inicio:
14/10/2024Fecha prevista finalización:
31/12/2024
La AEMPS ha autorizado unidades por comercialización excepcional. Disponibles unidades etiquetadas en un idioma diferente al castellano.
- Env. con 5 jeringas precargadas de 1 ml
- H: Medicamento de uso hospitalario
- Dispensación sujeta a prescripción médica
- No sustit.: Medicamento NO sustituible por el farmacéutico (Biológicos)
- Fi: Medicamento financiado sólo para determinadas indicaciones
- Facturable SNS: NO
- Comercializado: No
- Situación: Alta
- Código Nacional: 762406
- EAN13: 8470007624066
- Conservar en frío: Sí
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.MEDICAMENTO SUJETO A SEGUIMIENTO ADICIONAL. ATENCIóN EN FARMACOVIGILANCIA
 Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Beyfortus 50 mg solución inyectable en jeringa precargada,
Beyfortus 100 mg solución inyectable en jeringa precargada
2. COMPOSICIóN CUALITATIVA Y CUANTITATIVA
Beyfortus 50 mg solución inyectable en jeringa precargada:
Cada jeringa precargada contiene 50 mg de nirsevimab en 0,5 ml (100 mg/ml).
Beyfortus 100 mg solución inyectable en jeringa precargada:
Cada jeringa precargada contiene 100 mg de nirsevimab en 1 ml (100 mg/ml).
Nirsevimab es un anticuerpo monoclonal humano de inmunoglobulina G1 kappa (IgG1κ) producido en células de ovario de hámster chino (CHO) mediante tecnología de ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACéUTICA
Solución inyectable (inyectable).
Solución transparente a opalescente, incolora a amarilla, pH 6.0.
4. DATOS CLíNICOS
4.1 Indicaciones terapéuticas
Beyfortus está indicado para la prevención de la enfermedad de las vías respiratorias inferiores producida por el Virus Respiratorio Sincitial (VRS) en neonatos y lactantes durante su primera temporada del VRS.
Beyfortus se debe usar de acuerdo con las recomendaciones oficiales.
4.2 Posología y forma de administración
Posología:
La dosis recomendada es una dosis única de 50 mg administrados vía intramuscular para lactantes con peso corporal <5 kg y una dosis única de 100 mg administrados vía intramuscular para lactantes con peso corporal ≥5 kg.
Beyfortus se debe administrar antes del comienzo de la temporada del VRS, o desde el nacimiento en lactantes nacidos durante la temporada del VRS.
La dosificación en lactantes con un peso corporal de 1,0 kg a <1,6 kg se basa en la extrapolación, no se dispone de datos clínicos. Se prevé que la exposición en lactantes de <1 kg de peso sea mayor que en aquellos que pesan más. Se deben considerar cuidadosamente los beneficios y los riesgos del uso de nirsevimab en lactantes de <1 kg de peso.
Se dispone de datos limitados en lactantes extremadamente prematuros (edad gestacional [EG] <29 semanas) de menos de 8 semanas de edad. No se dispone de datos clínicos en lactantes con una edad posmenstrual (edad gestacional al nacimiento más edad cronológica) de menos de 32 semanas (ver sección 5.1).
En lactantes sometidos a cirugía cardíaca con bypass cardiopulmonar, se puede administrar una dosis adicional lo antes posible una vez que el lactante esté estable después de la cirugía para asegurar niveles séricos adecuados de nirsevimab. En los primeros 90 días tras recibir la primera dosis de Beyfortus, la dosis adicional debe ser de 50 mg o 100 mg según el peso corporal. Si han transcurrido más de 90 días después de la primera dosis, la dosis adicional podría ser una dosis única de 50 mg independientemente del peso corporal, para cubrir el resto de la temporada del VRS.
No hay datos de seguridad y eficacia disponibles en dosis repetidas.
No se ha establecido la seguridad y eficacia de nirsevimab en niños de 2 a 18 años.
No se dispone de datos.
Forma de administración:
Beyfortus se administra únicamente mediante inyección intramuscular.
Se administra por vía intramuscular, preferiblemente en la cara anterolateral del muslo. El músculo glúteo no se debe utilizar de forma rutinaria como lugar de inyección ya que existe el riesgo de dañar el nervio ciático.
Instrucciones de administración:
Beyfortus está disponible en una jeringa precargada de 50 mg y de 100 mg. Revise las etiquetas del envase y de la jeringa precargada para asegurarse de que ha seleccionado la presentación correcta de 50 mg o 100 mg según sea necesario.
| Beyfortus 50 mg (50 mg/0,5 ml) jeringa precargada con varilla de émbolo morado. | Beyfortus 100 mg (100 mg/1 ml) jeringa precargada con varilla de émbolo azul claro. |
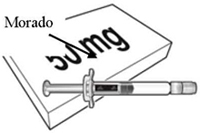 |  |
Consulte la Figura 1 para los componentes de la jeringa precargada.
Figura 1: Componentes de la jeringa con bloqueo Luer
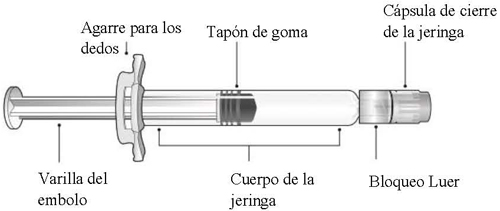
Paso 1: Sosteniendo el bloqueo Luer en una mano (evite sostener la varilla del émbolo o el cuerpo de la jeringa), desenrosque la cápsula de cierre de la jeringa girándola con la otra mano en sentido contrario a las agujas del reloj.
Paso 2: Coloque una aguja de bloqueo Luer a la jeringa precargada girando suavemente la aguja en el sentido de las agujas del reloj sobre la jeringa precargada hasta que se note una ligera resistencia.
Paso 3: Sostenga el cuerpo de la jeringa con una mano y tire con cuidado del capuchón de la aguja directamente con la otra mano. No sostenga la varilla del émbolo mientras retira la cubierta de la aguja o el tapón de goma podría moverse. No toque la aguja ni deje que toque ninguna superficie. No vuelva a tapar la aguja ni la desprenda de la jeringa.
Paso 4: Administre todo el contenido de la jeringa precargada como inyección intramuscular, preferiblemente en la cara anterolateral del muslo. El músculo glúteo no se debe utilizar rutinariamente como sitio de inyección debido al riesgo de daño al nervio ciático.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Trazabilidad:
Con objeto de mejorar la trazabilidad de los medicamentos biológicos, el nombre y el número de lote del medicamento administrado deben estar claramente registrados.
Hipersensibilidad incluyendo anafilaxia:
Se han notificado reacciones graves de hipersensibilidad, incluida la anafilaxia, con anticuerpos monoclonales. Si se observan signos y síntomas de una reacción de hipersensibilidad o anafilaxia clínicamente significativa, suspenda inmediatamente la administración e inicie el tratamiento adecuado con medicamentos y/o terapia de soporte.
Trastornos hemorrágicos clínicamente significativos
Al igual que con otras inyecciones intramusculares, nirsevimab se debe administrar con precaución en lactantes con trombocitopenia o cualquier trastorno de la coagulación.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones. Los anticuerpos monoclonales no suelen tener un potencial significativo de interacción, ya que no afectan directamente a las enzimas del citocromo P450 y no son sustratos de transportadores hepáticos o renales. Los efectos indirectos en las enzimas del citocromo P450 son poco probables ya que la diana de nirsevimab es un virus exógeno.
Administración concomitante con vacunas:
Dado que nirsevimab es un anticuerpo monoclonal, no se espera que una inmunización pasiva específica del VRS interfiera en la respuesta inmune activa de las vacunas coadministradas.
La experiencia en la coadministración con vacunas es limitada. En ensayos clínicos, cuando nirsevimab se administró con vacunas infantiles habituales, el perfil de seguridad y reactogenicidad del régimen coadministrado fue similar al de las vacunas infantiles administradas de forma aislada. Nirsevimab se puede administrar concomitantemente con vacunas infantiles.
Nirsevimab no se debe mezclar con ninguna vacuna en la misma jeringa o vial (ver sección 6.2). Cuando se administra concomitantemente con vacunas inyectables, se debe administrar con jeringas separadas y en diferentes lugares de inyección.
4.6 Fertilidad, embarazo y lactancia
No procede.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No procede.
4.8 Reacciones adversas
Resumen del perfil de seguridad:
La reacción adversa más frecuente fue la erupción (0,7%) que se produjo en los 14 días posteriores a la dosis. La mayoría de los casos fueron de intensidad leve a moderada. Además, se notificaron pirexia y reacciones en el lugar de la inyección en una tasa de 0,5% y 0,3% dentro de los 7 días posteriores a la dosis, respectivamente. Las reacciones en el lugar de la inyección no fueron graves.
Tabla de reacciones adversas
La Tabla 1 presenta las reacciones adversas notificadas en 2 966 lactantes nacidos a término y prematuros (EG ≥29 semanas) que recibieron nirsevimab en ensayos clínicos.
Las reacciones adversas notificadas en ensayos clínicos controlados se clasifican según la clasificación por órganos y sistemas (COS) de MedDRA. Dentro de cada COS, los términos preferentes se presentan en orden decreciente de frecuencia y después en orden decreciente de gravedad. Las frecuencias de aparición de reacciones adversas se definen como: muy frecuentes (≥1/10); frecuentes (≥1/100 a <1/10); poco frecuentes (≥1/1 000 a <1/100); raras (≥1/10 000 a <1/1 000); muy raras (<1/10 000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas
| MedDRA COS | Término preferente de MedDRA | Frecuencia |
| Trastornos de la piel y del tejido subcutáneo | Erupcióna | Poco frecuentes |
| Trastornos generales y alteraciones en el lugar de administración | Reacción en el lugar de la inyecciónb | Poco frecuentes |
| Pirexia | Poco frecuentes |
a La erupción se definió mediante los siguientes términos preferentes agrupados: erupción cutánea, erupción maculopapular, erupción macular.
b La reacción en el lugar de la inyección se definió mediante los siguientes términos preferentes agrupados: reacción en el lugar de la inyección, dolor en el lugar de la inyección, inflamación en el lugar de la inyección, edema en el lugar de la inyección, inflamación en el lugar de la inyección.
Lactantes con mayor riesgo de padecer la enfermedad grave por VRS:
La seguridad también se evaluó en MEDLEY en 918 lactantes con mayor riesgo de padecer la enfermedad grave por VRS, incluidos 196 lactantes extremadamente prematuros (EG <29 semanas) y 306 lactantes con enfermedad pulmonar crónica de la prematuridad, o cardiopatía congénita hemodinámicamente significativa, que recibieron nirsevimab (614) o palivizumab (304) entrando en su primera temporada del VRS. El perfil de seguridad fue comparable al del comparador palivizumab y consistente con el perfil de seguridad en lactantes a término y prematuros EG ≥29 semanas (D5290C00003 y MELODY).
Inmunogenicidad:
Al igual que con todas las proteínas terapéuticas, existe un potencial de inmunogenicidad.
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es.
4.9 Sobredosis
No hay ningún tratamiento específico en caso de sobredosis con nirsevimab. Si se produce sobredosis, se debe monitorizar al paciente para detectar la aparición de reacciones adversas y se le debe proporcionar tratamiento sintomático según sea necesario.
5. PROPIEDADES FARMACOLóGICAS
5.1 Propiedades farmacodinámicas
5.2 Propiedades farmacocinéticas
5.3 Datos preclínicos sobre seguridad
6.DATOS FARMACéUTICOS
6.1 Lista de excipientes
L-histidina,
Hidrocloruro de L-histidina,
Hidrocloruro de L-arginina,
Sacarosa,
Polisorbato 80,
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
2 años.
Beyfortus se puede mantener a temperatura ambiente (20°C - 25°C) protegido de la luz durante un máximo de 8 horas. Después de este tiempo, la jeringa se debe desechar.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
No agitar ni exponer al calor directo.
Conservar la jeringa precargada en el embalaje exterior para protegerla de la luz.
Para condiciones de conservación del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Jeringa precargada de vidrio tipo I con bloqueo Luer siliconado con un tapón de émbolo recubierto de FluroTec.
Cada jeringa precargada contiene 0,5 ml o 1 ml de solución.
Tamaños de envases:
- 1 o 5 jeringa(s) precargada(s) sin agujas.
- 1 jeringa precargada envasada con dos agujas separadas de diferentes tamaños.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Este medicamento debe administrarse por un profesional sanitario entrenado usando técnicas asépticas para asegurar la esterilidad.
Inspeccione visualmente el medicamento para detectar partículas y decoloración antes de la administración. El medicamento es una solución transparente a opalescente, incolora a amarilla. No inyecte si el líquido está turbio, decolorado o contiene partículas grandes o partículas extrañas.
No lo utilice si la jeringa precargada se ha caído o dañado o si se ha roto el sello de seguridad del envase.
Eliminación:
Cada jeringa precargada es para un solo uso. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIóN DE COMERCIALIZACIóN
AstraZeneca AB,
SE151 85 Södertälje,
Suecia
8. NúMERO(S) DE AUTORIZACIóN DE COMERCIALIZACIóN
| EU/1/22/1689/001 | 50 mg, 1 jeringa precargada de un solo uso |
| EU/1/22/1689/003 | 50 mg, 5 jeringas precargadas de un solo uso |
| EU/1/22/1689/004 | 100 mg, 1 jeringa precargada de un solo uso |
| EU/1/22/1689/006 | 100 mg, 5 jeringas precargadas de un solo uso |
9. FECHA DE LA PRIMERA AUTORIZACIóN/RENOVACIóN DE LA AUTORIZACIóN
Fecha de la primera autorización: 31 octubre 2022
10. FECHA DE LA REVISIóN DEL TEXTO
Junio 2023
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
11.PRESENTACIóN, PRECIO Y CONDICIONES DE PRESCRIPCIóN Y DISPENSACIóN:
BEYFORTUS 50 MG SOLUCION INYECTABLE EN JERINGA PRECARGADA, 1 jeringa precargada (CN: 762403) PVP notificado: 812,67 € PVP IVA notificado: 845,18€. Financiada por el SNS para las indicaciones terapéuticas autorizadas según las recomendaciones oficiales que publica periódicamente el Ministerio de Sanidad. Con receta.
Puede acceder a información detallada y actualizada sobre este medicamento escaneando con su teléfono móvil (smartphone) el código QR

BEYFORTUS 100 MG SOLUCION INYECTABLE EN JERINGA PRECARGADA, 1 jeringa precargada (CN: 762405) PVP notificado: 812,67 € PVP IVA notificado: 845,18 €. Financiada por el SNS para las indicaciones terapéuticas autorizadas según las recomendaciones oficiales que publica periódicamente el Ministerio de Sanidad. Con receta.
Puede acceder a información detallada y actualizada sobre este medicamento escaneando con su teléfono móvil (smartphone) el código QR

CONSULTE LA FICHA TéCNICA COMPLETA ANTES DE PRESCRIBIR ESTE MEDICAMENTO.

